5. 应用案例¶
5.1. CdTe的本征缺陷计算¶
5.1.1. 准备计算PREPARE¶
5.1.1.1. 准备POSCAR与dasp.in¶
Materials Project 数据库中找到CdTe原胞的POSCAR结构,显示如下:Cd1 Te1
1.0
4.6874446869 0.0000000000 0.0000000000
2.3437223434 4.0594461777 0.0000000000
2.3437223434 1.3531487259 3.8272825601
Cd Te
1 1
Direct
0.000000000 0.000000000 0.000000000
0.750000000 0.750000000 0.750000000
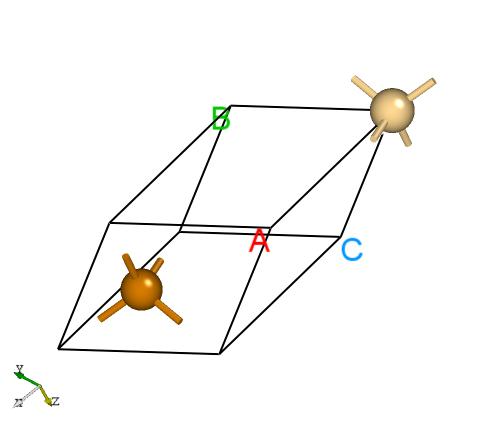
CdTe的原胞结构。
dasp.in 中写入必要参数############## Job Scheduling ##############
cluster = SLURM # (job scheduling system)
node_number = 2 # (number of node)
core_per_node = 52 # (core per node)
queue = batch # (name of queue/partition)
max_time = 24:00:00 # (maximum time for a single DFT calculation)
vasp_path_dec = /opt/vasp.5.4.4/bin/vasp_gam # (path of VASP)
vasp_path_tsc = /opt/vasp.5.4.4/bin/vasp_std
job_name = submit_job # (name of script)
potcar_path = /opt/POT/potpaw_PBE # (path of pseudopotentials)
max_job = 5
############## TSC Module ##############
database_api = ******************* # (str-list type)
############## DEC Module ##############
level = 2 # (level=1: PBE+PBE; level=2: PBE+HSE; level=3: HSE+HSE)
min_atom = 190
max_atom = 240
intrinsic = T # (default: T)
correction = FNV # (default: none)
epsilon = 10.3
Eg_real = 1.45 # (experimental band gap)
############## DDC Module ##############
ddc_temperature = 1000 300
ddc_mass = 0.09 0.84
dasp.in 中所有列出的参数进行说明。cluster = SLURM
# 表示使用集群的队列系统为SLURM
node_number = 2
# 对于每个单独的计算,使用2个节点
core_per_node = 52
# 对于每个节点,使用52个核。因此对于每个单独的计算,总共使用2*52=104核
queue = batch
# 使用名为"batch"的队列进行计算。因此,在设置dasp.in参数之前,需要确认超算/集群上的队列名、节点、核数
max_time = 24:00:00 # (maximum time for a single DFT calculation)
# 每个单独的计算所允许的最大时间为24小时,可任意设置。
vasp_path_dec = /opt/vasp.5.4.4/bin/vasp_gam # (path of VASP)
vasp_path_tsc = /opt/vasp.5.4.4/bin/vasp_std
# 对于TSC的计算,采用std版的VASP。对于DEC的单k点计算,采用gam版的VASP。
job_name = submit_job # (name of script)
# 提交任务的脚本,命名为"submit_job",可任意设置。
potcar_path = /opt/POT/potpaw_PBE # (path of pseudopotentials)
# POTCAR的路径
max_job = 5
# 允许同时在跑的任务最大数
database_api = ******************* # (str-list type)
# 用于访问Materials Project数据库
level = 2 # (level=1: PBE+PBE; level=2: PBE+HSE; level=3: HSE+HSE)
# 对于超胞,使用PBE优化其原子位置,使用HSE计算其总能
min_atom = 190
max_atom = 240
# 我们希望生成的超胞大小在190-240个原子之间,且尽量使a=b=c,a⊥b⊥c
intrinsic = T # (default: T)
# 产生本征缺陷,V_Cd V_Te Cd_Te Te_Cd Cd_i Te_i
correction = FNV # (default: none)
# 带电缺陷的修正方案采用FNV修正
epsilon = 10.3
# CdTe的介电常数为10.3
Eg_real = 1.45 # (experimental band gap)
# CdTe的实验带隙值约为1.45 eV,DASP将根据此数据调整AEXX参数,从而使得无缺陷超胞的带隙值等于1.45 eV
ddc_temperature = 1000 300
# 设置生长温度为1000 K,工作温度为300 K
ddc_mass = 0.09 0.84
# 设置电子有效质量为0.09,空穴有效质量为0.84
5.1.1.2. 使用DASP产生必要输入文件¶
dasp.in 文件,执行 dasp 1 ,即可启动PREPARE模块,此后无需额外操作。DASP会输出 1prepare.out 文件记录程序的运行日志。5.1.1.3. PREPARE模块运行流程¶
产生超胞:
POSCAR_nearlycube :Cubic_cell
1.0
19.8871435472 0.0000000000 0.0000000000
0.0000000000 19.8871435472 0.0000000000
0.0000000000 0.0000000000 19.8871435472
Cd Te
108 108
Direct
0.0000000000 0.0000000000 0.0000000000
0.8333333333 1.0000000000 0.1666666666
0.8333333333 0.1666666666 1.0000000000
0.6666666666 0.1666666666 0.1666666666
...
通过可视化软件,我们可以看到:给出的CdTe原胞晶轴的夹角是较小的,但是经过DASP生成的超胞是三边垂直的。
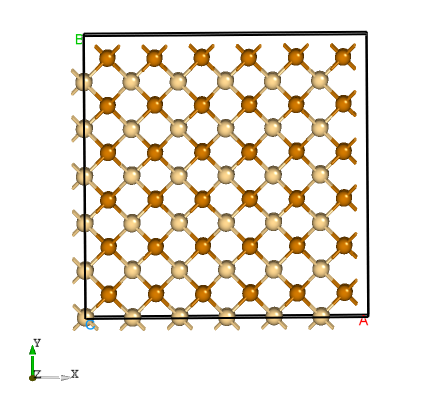
CdTe的超胞结构。
马德隆常数计算:
1prepare.out 的输出如下:############ Prepare Files module start ############
Read the structure file POSCAR you provided
Get the refined cell POSCAR_refined from POSCAR
Generate the nearlycube cell POSCAR_nearlycube from POSCAR
Generate job script through dasp.in parameters
Generate single-point KPOINTS
Generate pseudopotential file POTCAR through potcar_dir you set
Generate commonly used vasp input file INCAR
Start the madelung constant calculation
Generate the madelung calculation directory
Generate madelung calculation POSCAR
Generate madelung calculation POTCAR
Generate madelung calculation INCAR
Generate madelung calculation KPOINTS
Generate madelung calculation job script
Job 103.host5 submitted: /home/test/CdTe/dec/madelung/static
Succeed job 103.host5: /home/test/CdTe/dec/madelung/static
The madelung constant calculation completed
The madelung constant = 2.837
HSE交换参数计算:
cd ./dec/AEXX
ls
0.25 0.25880073638027207 0.3 AEXX.list
1prepare.out 可以看到如下日志:Start the HSE parameter AEXX calculation
Job 107.host5 submitted: /home/test/CdTe/dec/AEXX/0.25/static
Job 108.host5 submitted: /home/test/CdTe/dec/AEXX/0.3/static
Succeed job 107.host5: /home/test/CdTe/dec/AEXX/0.25/static
Succeed job 108.host5: /home/test/CdTe/dec/AEXX/0.3/static
Job 108.host5 submitted: /home/test/CdTe/dec/AEXX/0.25880073638027207/static
Succeed job 108.host5: /home/test/CdTe/dec/AEXX/0.25880073638027207/static
The HSE parameter AEXX calculation completed
The HSE parameter AEXX = 0.26
level = 2: Generate PBE relax vasp input file INCAR-relax
level = 2: Generate HSE static vasp input file INCAR-static
host超胞原子位置的优化:
POSCAR_final 。同时也可以在 1prepare.out 可以DASP运行结束的标志,并告诉我们下一步需要做TSC模块的计算。Start the POSCAR_nearlycube relax calculation
Generate the POSCAR_nearlycube relax directory
Job 109.host5 submitted: /home/test/CdTe/dec/relax
Succeed job 109.host5: /home/test/CdTe/dec/relax
The POSCAR_nearlycube relax calculation completed
Get the final structure POSCAR_final
############ Prepare Files module end ############
PREPARE模块 finished, please run DASP-TSC next
5.1.2. 热力学稳定性和元素化学势计算TSC¶
5.1.2.1. 运行TSC模块¶
dasp 1 执行PREPARE模块时,会生成CdTe/dec目录,并在该目录中产生 1prepare.out 文件。等待程序执行完毕, 1prepare.out 有相应的完成标志。进入CdTe/dec目录。确认INCAR-relax,INCAR-static文件中的参数是可行的。(用户可修改INCAR,DASP将根据此目录中的INCAR做后续的计算)dasp 2 执行TSC模块。同样地,TSC模块会在CdTe目录中生成名为tsc的目录,里面记录了TSC程序的计算输出,包括各计算目录以及运行日志文件 2tsc.out 。等待程序完成期间无需额外操作。5.1.2.2. TSC模块运行流程¶
host结构的总能计算(与MP参数保持一致):
Materials Project 数据库完全一致的输入参数(INCAR,KPOINTS,POTCAR)来对用户给定的原胞做结构优化和静态计算。因此,该计算得到的总能与MP数据库的总能是可比的。此步骤是为了得到影响CdTe稳定性的 关键杂相 。通过目录可以看到:cd tsc
cd CdTe/
ls
relaxation1 relaxation2 static
关键杂相判断:
2tsc.out 中可看到相关的信息:...
analysing the thermodynamic stability of CdTe.
key phases of CdTe are: Cd Te .
file key_phases_info_recalc.yaml generated.
analysing of CdTe is done.
...
host与杂相结构的总能计算(PREPARE模块确定的参数):
2tsc.out 如下:...
Job 112.host5 submitted: /home/test/CdTe/tsc/CdTe/static_recalc
Job 113.host5 submitted: /home/test/CdTe/tsc/Cd/static_recalc
Job 114.host5 submitted: /home/test/CdTe/tsc/Te/static_recalc
Succeed job 112.host5: /home/test/CdTe/tsc/CdTe/static_recalc
Succeed job 113.host5: /home/test/CdTe/tsc/Cd/static_recalc
Succeed job 114.host5: /home/test/CdTe/tsc/Te/static_recalc
...
化学势的计算:
dasp.in :# 顺序与POSCAR中元素顺序一致,即第一列是Cd,第二列是Te
E_pure = -1.7736 -4.6974
p1 = 0.0 -1.1854
p2 = -1.1854 0.0
2tsc.out 可以看到程序执行完毕的输出:dir '2d-figures','3d-figures','ori_data_MP' ready. try to read file: 'calc_list.yaml'.
analysing the thermodynamic stability of CdTe.
key phases of CdTe are: Cd Te .
analysing of CdTe is done.
DASP-TSC finished
5.1.3. 缺陷形成能和转变能级计算DEC¶
5.1.3.1. 运行DEC模块¶
dasp 2 执行TSC模块时,会生成CdTe/tsc目录,并在该目录中产生 2tsc.out 文件。等待程序执行完毕, 2tsc.out 有相应的完成标志。打开CdTe/dasp.in,确认化学势已被程序自动输入。dasp 3 执行DEC模块。DEC会在第一步已经生成的dec目录中继续输出相关文件,包括缺陷结构,缺陷目录,以及运行日志文件 3dec.out 。等待程序完成期间无需额外操作。5.1.3.2. DEC模块运行流程¶
产生缺陷结构:
dasp.in 中的参数intrinsic = T,DEC模块将产生CdTe的本征缺陷,即生成CdTe/dec/Intrinsic_Defect计算目录,在其下面分别有空位缺陷V_Cd,V_Te,反位缺陷Cd_Te,Te_Cd,间隙位缺陷Cd_i,Te_i的缺陷结构和目录。根据对称性判断,CdTe晶格中不存在非等价的Cd和Te原子,因此同样的缺陷构型只需产生一种。cd dec/Intrinsic_Defect/
ls
Cd_i Cd_Te1 host Intrinsic_Defect.list Te_Cd1 Te_i V_Cd1 V_Te1
3dec.out 看到DEC模块的输出如下:############ Neutral Defect module start ############
Make intrinsic defect directory Intrinsic_Defect
Generate host directory in Intrinsic_Defect
Start generating neutral vacancy defect
Generate neutral defect at: V_Cd1/initial_structure/q0
Generate neutral defect at: V_Te1/initial_structure/q0
Neutral vacancy defect generation completed
Start generating neutral intrinsic antisite defect
Generate neutral defect at: Te_Cd1/initial_structure/q0
Generate neutral defect at: Cd_Te1/initial_structure/q0
Neutral intrinsic antisite defect generation completed
Start generating neutral intrinsic interstitial defect
Generate neutral defect at: Cd_i/random1/initial_structure/q0
Generate neutral defect at: Cd_i/random2/initial_structure/q0
Generate neutral defect at: Cd_i/random3/initial_structure/q0
Generate neutral defect at: Cd_i/random4/initial_structure/q0
Generate neutral defect at: Cd_i/random5/initial_structure/q0
Generate neutral defect at: Cd_i/random6/initial_structure/q0
Generate neutral defect at: Te_i/random1/initial_structure/q0
Generate neutral defect at: Te_i/random2/initial_structure/q0
Generate neutral defect at: Te_i/random3/initial_structure/q0
Generate neutral defect at: Te_i/random4/initial_structure/q0
Generate neutral defect at: Te_i/random5/initial_structure/q0
Generate neutral defect at: Te_i/random6/initial_structure/q0
Neutral intrinsic interstitial defect generation completed
############ Neutral Defect module end ############
提交各缺陷q=0计算任务:
dasp.in 中 level = 2 的参数),此步骤等待时间较长。可随时检查 3dec.out 文件。 3dec.out 中的相关信息如下所示:Job 245.host5 submitted: /home/test/CdTe/dec/Intrinsic_Defect/Cd_Te1/initial_structure/q0
Job 246.host5 submitted: /home/test/CdTe/dec/Intrinsic_Defect/Te_Cd1/initial_structure/q0
Job 247.host5 submitted: /home/test/CdTe/dec/Intrinsic_Defect/V_Te1/initial_structure/q0
Job 248.host5 submitted: /home/test/CdTe/dec/Intrinsic_Defect/Te_i/random3/initial_structure/q0
Job 249.host5 submitted: /home/test/CdTe/dec/Intrinsic_Defect/Te_i/random2/initial_structure/q0
Failed job 245.host5: /home/test/CdTe/dec/Intrinsic_Defect/Cd_Te1/initial_structure/q0
...
产生带电缺陷的计算目录:
3dec.out 中的相关信息如下所示:############ Ionized Defect module start ############
Start generating ionized defects
Warning: no EIGENVAL in /home/test/CdTe/dec/Intrinsic_Defect/Cd_Te1/initial_structure/q0/static, skipped this directory!
Start generating ionized defects
Ionized defect path: /home/test/CdTe/dec/Intrinsic_Defect/V_Te1/initial_structure/q+1
Ionized defect path: /home/test/CdTe/dec/Intrinsic_Defect/V_Te1/initial_structure/q+2
Ionized defects generation completed
Start generating ionized defects
Ionized defect path: /home/test/CdTe/dec/Intrinsic_Defect/Te_i/random3/initial_structure/q+1
Ionized defect path: /home/test/CdTe/dec/Intrinsic_Defect/Te_i/random3/initial_structure/q+2
Ionized defect path: /home/test/CdTe/dec/Intrinsic_Defect/Te_i/random3/initial_structure/q+3
Ionized defect path: /home/test/CdTe/dec/Intrinsic_Defect/Te_i/random3/initial_structure/q+4
...
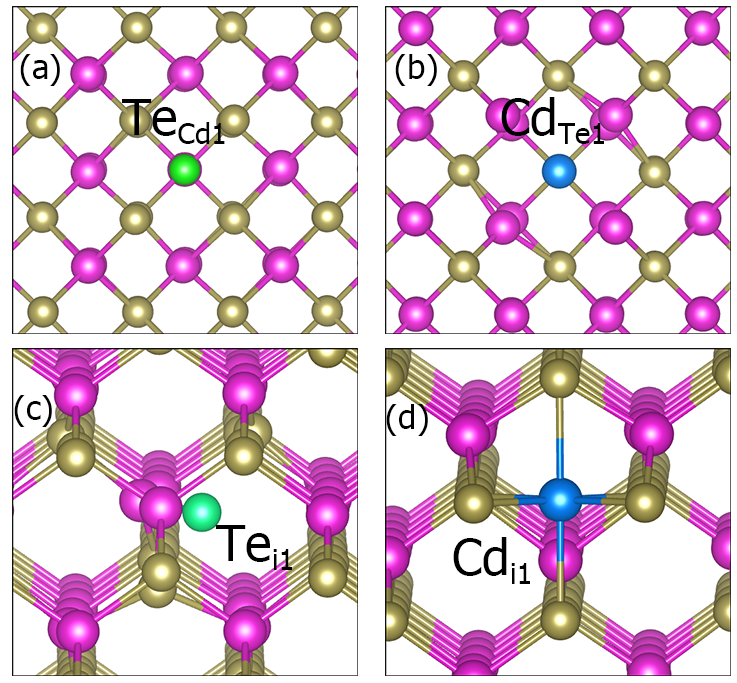
CdTe的部分缺陷结构。
提交各缺陷q≠0的计算任务:
dasp.in 中 level = 2 的参数),此步骤的等待时间比 q=0 计算时间更长。 3dec.out 中的相关信息如下所示:############ AutoRun - Ionized Defect module start ############
Job 693.host5 submitted: /home/test/CdTe/dec/Intrinsic_Defect/V_Te1/initial_structure/q+2
Job 694.host5 submitted: /home/test/CdTe/dec/Intrinsic_Defect/V_Te1/initial_structure/q+1
Job 695.host5 submitted: /home/test/CdTe/dec/Intrinsic_Defect/Te_i/random3/initial_structure/q+2
Job 696.host5 submitted: /home/test/CdTe/dec/Intrinsic_Defect/Te_i/random3/initial_structure/q+1
Job 700.host5 submitted: /home/test/CdTe/dec/Intrinsic_Defect/Te_i/random3/initial_structure/q+4
Succeed job 694.host5: /home/test/CdTe/dec/Intrinsic_Defect/V_Te1/initial_structure/q+1
Succeed job 693.host5: /home/test/CdTe/dec/Intrinsic_Defect/V_Te1/initial_structure/q+2
...
计算带电缺陷的修正:
dasp.in 中 correction = FNV 的参数),并计算其缺陷形成能和转变能级。每个缺陷各价态的修正量和形成能的具体数值,都记录在 3dec.out 中:...
The formation energy (neutral) of V_Te1 at p1 is 1.684321
The formation energy (neutral) of V_Te1 at p2 is 2.869721
The FNV correction (q = 2) E_correct = 0.279795 eV
The transition level (0/2+) above VBM: 1.2429
The FNV correction (q = 1) E_correct = 0.087991 eV
The transition level (0/+) above VBM: 1.2833
...
defect.log 文件中。输出形成能图像:
redo.in 的文件,在里面写入/home/test/CdTe/dec/Intrinsic_Defect/Cd_Te1/initial_structure/q0。dasp 3 执行DEC模块。程序会自动判断已经完成的计算,并根据 redo.in 重新计算该缺陷。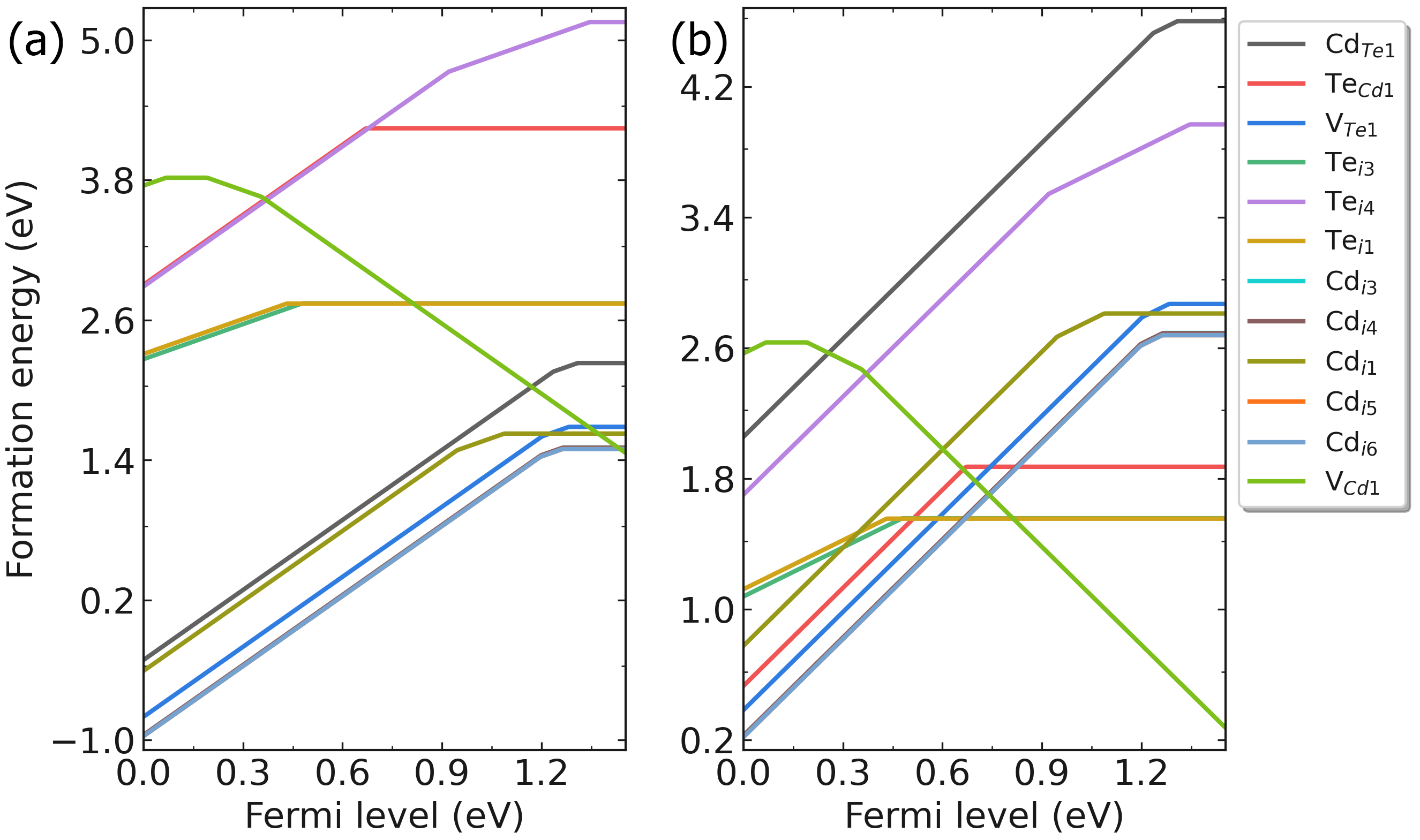
CdTe在(a)富Cd和(b)富Te时的缺陷形成能随费米能级的变化。
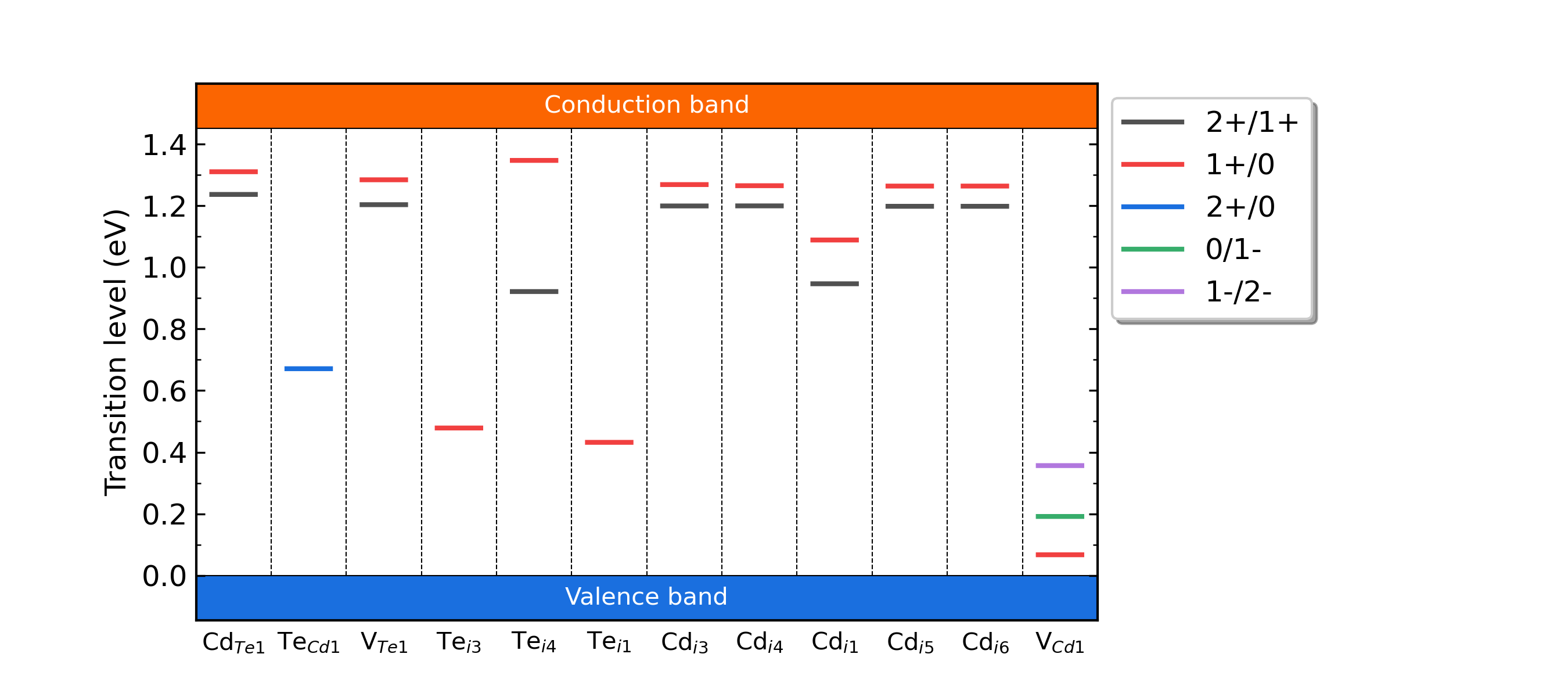
CdTe的缺陷转变能级。
5.1.4. 缺陷浓度和费米能级计算DDC¶
5.1.4.1. 运行DDC模块¶
dasp 4 执行DDC模块。等待期间无需额外操作。5.1.4.2. DDC模块运行流程¶
DefectParams.txt 文件中。4ddc.out :############ Collecting information from DEC ############
Read defect types from DEC calculation successfully.
Defects considered in DDC calculation: ['Cd_Te1', 'Te_Cd1', 'V_Te1', 'Te_i-3', 'Te_i-4', 'Te_i-1', 'Cd_i-3', 'Cd_i-4', 'Cd_i-1', 'Cd_i-5', 'Cd_i-6', 'V_Cd1']
Chemical potentials change from p1 to p2.
Calculate gq for defect in each charge state.
Calculate Nsites for Cd_Te1: 1.373114e+22 cm^-3.
Calculate Nsites for Te_Cd1: 1.373114e+22 cm^-3.
Calculate Nsites for V_Te1: 1.373114e+22 cm^-3.
Calculate Nsites for Te_i-3: 9.154092e+21 cm^-3.
Calculate Nsites for Te_i-4: 9.154092e+21 cm^-3.
Calculate Nsites for Te_i-1: 9.154092e+21 cm^-3.
Calculate Nsites for Cd_i-3: 9.154092e+21 cm^-3.
Calculate Nsites for Cd_i-4: 9.154092e+21 cm^-3.
Calculate Nsites for Cd_i-1: 9.154092e+21 cm^-3.
Calculate Nsites for Cd_i-5: 9.154092e+21 cm^-3.
Calculate Nsites for Cd_i-6: 9.154092e+21 cm^-3.
Calculate Nsites for V_Cd1: 1.373114e+22 cm^-3.
############ Collecting information from DEC ############
DefectParams.txt 文件:1000 300
0.090000 0.840000
1.454001
Cd_Te1 1.373114e+22 1 1.3094 2 1.2726 1 x x x x x x x x x x x x 2.230998 4.601798
Te_Cd1 1.373114e+22 1 0.4662 2 0.6708 1 0.3386 2 0.1484 1 x x x x x x x x 4.243870 1.873070
V_Te1 1.373114e+22 1 1.2833 2 1.2429 1 x x x x x x x x x x x x 1.684321 2.869721
Te_i-3 9.154092e+21 1 0.4786 2 0.153 1 0.1435 2 0.012 1 x x x x x x x x 2.743050 1.557650
Te_i-4 9.154092e+21 1 1.3461 2 1.1335 1 0.6062 2 0.4609 1 x x x x x x x x 5.154681 3.969281
Te_i-1 9.154092e+21 1 0.432 2 0.1299 1 0.0022 2 -0.0831 1 x x x x x x x x 2.740914 1.555514
Cd_i-3 9.154092e+21 1 1.2677 2 1.2331 1 0.7237 2 0.4498 1 x x x x x x x x 1.506642 2.692042
Cd_i-4 9.154092e+21 1 1.2641 2 1.2314 1 0.7192 2 0.4466 1 x x x x x x x x 1.505396 2.690796
Cd_i-1 9.154092e+21 1 1.0881 2 1.0171 1 0.5865 2 0.3569 1 x x x x x x x x 1.626088 2.811488
Cd_i-5 9.154092e+21 1 1.2629 2 1.2301 1 0.7187 2 0.4461 1 x x x x x x x x 1.493426 2.678826
Cd_i-6 9.154092e+21 1 1.2629 2 1.2301 1 0.7187 2 0.4461 1 x x x x x x x x 1.493426 2.678826
V_Cd1 1.373114e+22 1 0.0676 2 0.0115 1 -0.0427 2 -0.0964 1 0.1918 2 0.2742 1 0.7753 2 1.0413 1 3.819933 2.634533
生长温度下自洽计算:
DDC模块在T=1000 K( ddc_temperature = 1000 300 )的时候计算其缺陷浓度和载流子浓度,并根据电中性条件自洽求解费米能级。
工作温度下自洽计算:
DDC模块在T=300 K( ddc_temperature = 1000 300 )的时候重新分布每个缺陷各价态的浓度,并根据电中性条件再次自洽求解费米能级。
输出缺陷浓度:
DDC模块在CdTe/ddc目录下,输出四个文件: Fermi.dat Carrier.dat Defect_charge.dat density.png 。分别采用不同的生长温度, density.png 的结果如下:
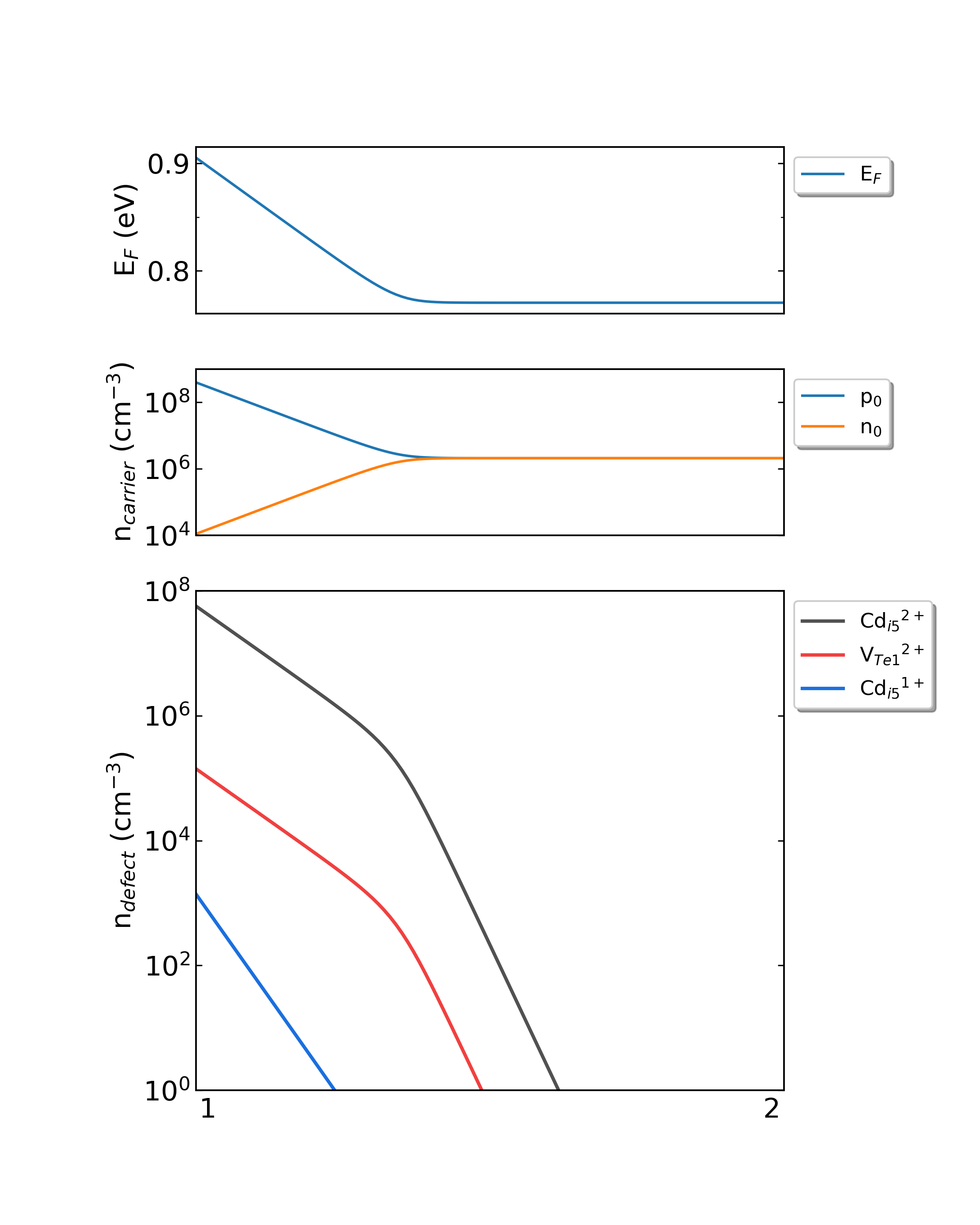
生长温度为300 K,工作温度为300 K时,CdTe的费米能级、载流子浓度和缺陷浓度随化学势的变化。
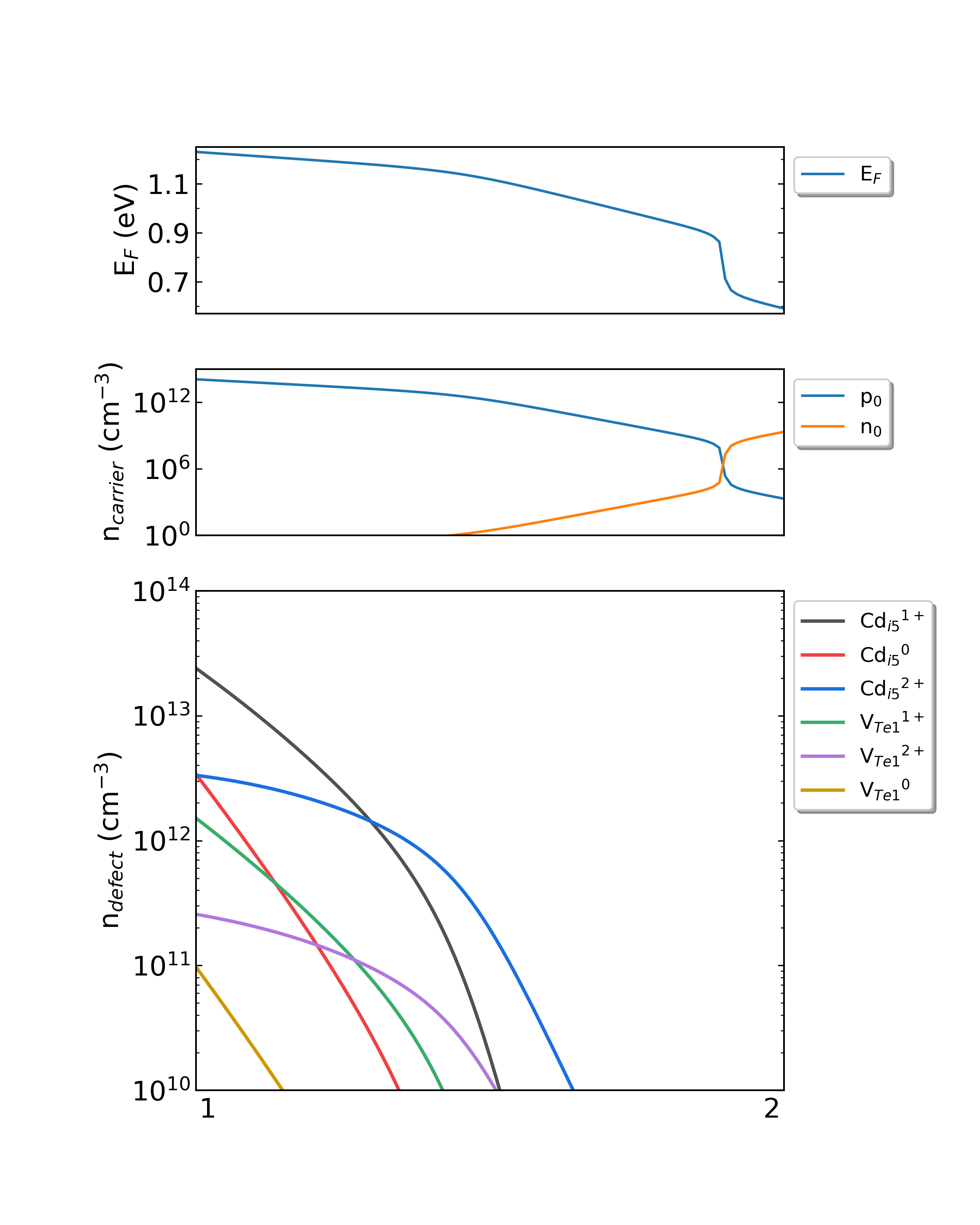
生长温度为600 K,工作温度为300 K时,CdTe的费米能级、载流子浓度和缺陷浓度随化学势的变化。
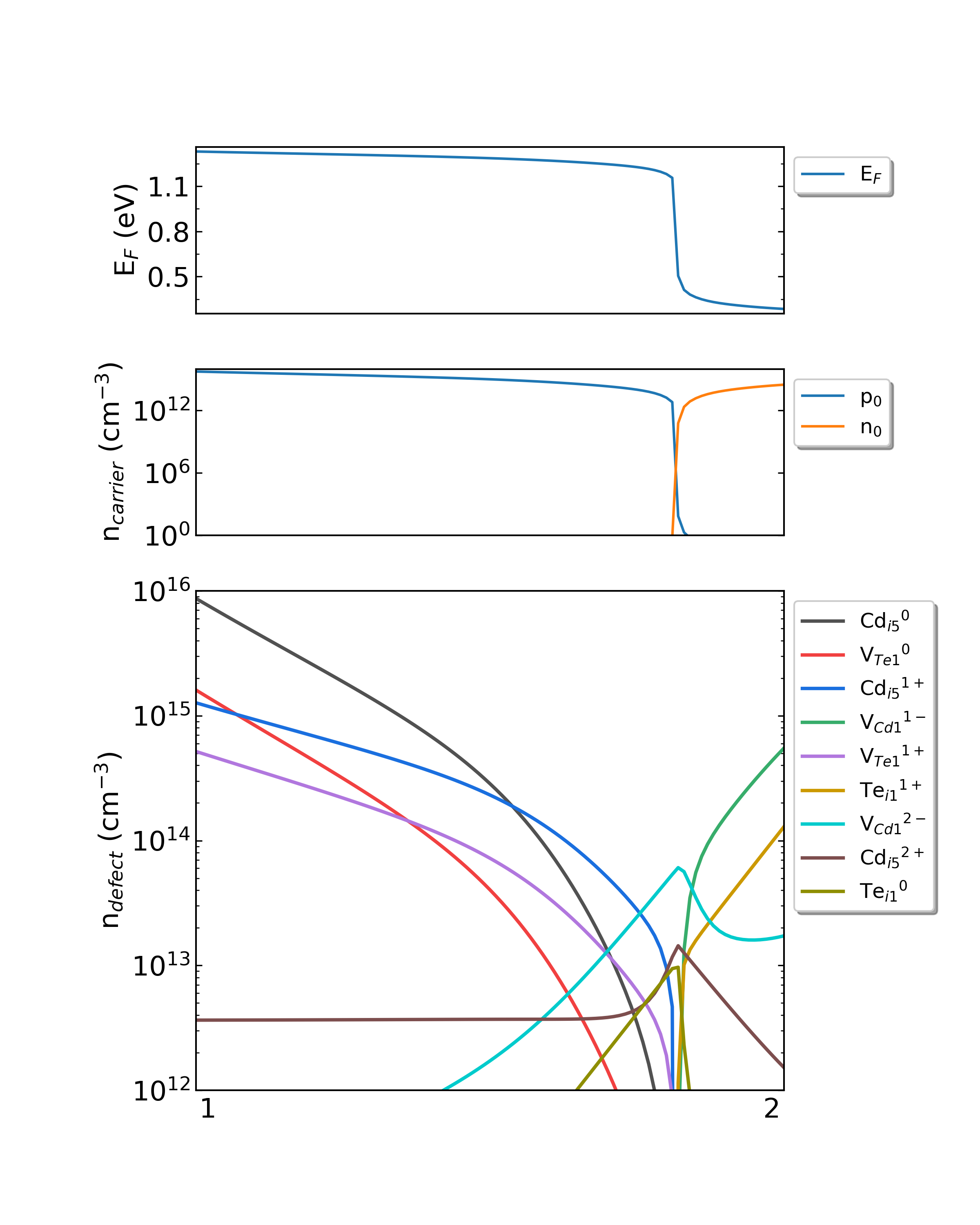
生长温度为1000 K,工作温度为300 K时,CdTe的费米能级、载流子浓度和缺陷浓度随化学势的变化。
5.2. HfO2的本征缺陷计算¶
5.2.1. 准备计算PREPARE¶
5.2.1.1. 准备POSCAR与dasp.in¶
Materials Project 数据库中找到HfO2的 POSCAR 文件,显示如下:Hf4 O8
1.00000000000000
5.0652456351012756 0.0000000000000000 -0.8648023964655065
0.0000000000000000 5.1942160710694010 0.0000000000000000
-0.0006123763055649 0.0000000000000000 5.3264852554835196
Hf O
4 8
Direct
0.7239198560286844 0.5430528338948646 0.2919471319794364
0.2760801439713155 0.0430528338948647 0.2080528680205637
0.2760801439713155 0.4569471661051352 0.7080528680205705
0.7239198560286844 0.9569471661051354 0.7919471319794295
0.5514108623083260 0.2575137054162841 0.0226462276199697
0.4485891376916739 0.7575137054162840 0.4773537723800304
0.4485891376916739 0.7424862945837160 0.9773537723800304
0.5514108623083260 0.2424862945837159 0.5226462276199696
0.9317881306845183 0.6693565882783469 0.6530354073676818
0.0682118693154819 0.1693565882783468 0.8469645926323182
0.0682118693154819 0.3306434117216532 0.3469645926323183
0.9317881306845183 0.8306434117216531 0.1530354073676817
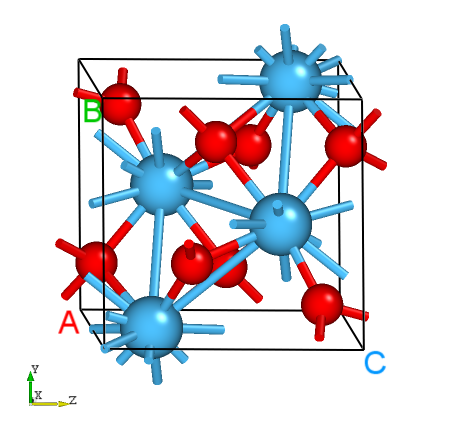
HfO2的晶体结构。
dasp.in 中写入必要参数############## Job Scheduling ##############
cluster = PBS # (job scheduling system)
node_number = 1 # (number of node)
core_per_node = 96 # (core per node)
queue = batch # (name of queue/partition)
max_time = 24:00:00 # (maximum time for a single DFT calculation)
vasp_path_dec = /opt/vasp.5.4.4/bin/vasp_gam # (path of VASP)
vasp_path_tsc = /opt/vasp.5.4.4/bin/vasp_std
job_name = submit_job # (name of script)
potcar_path = /opt/POT/potpaw_PBE # (path of pseudopotentials)
max_job = 5
############## TSC Module ##############
database_api = ******************* # (str-list type)
############## DEC Module ##############
level = 2 # (level=1: PBE+PBE; level=2: PBE+HSE; level=3: HSE+HSE)
min_atom = 96
max_atom = 96
intrinsic = T # (default: T)
correction = FNV # (default: none)
epsilon = 21.6
Eg_real = 5.68 # (experimental band gap)
############## DDC Module ##############
ddc_temperature = 1000 300
ddc_mass = 2.95 2.99
dasp.in 中所有列出的参数进行说明。cluster = PBS
# 表示使用集群的队列系统为PBS
node_number = 1
# 对于每个单独的计算,使用1个节点
core_per_node = 96
# 对于每个节点,使用96个核。因此对于每个单独的计算,总共使用1*96=96核
queue = batch
# 使用名为"batch"的队列进行计算。因此,在设置dasp.in参数之前,需要确认超算/集群上的队列名、节点、核数
max_time = 24:00:00 # (maximum time for a single DFT calculation)
# 每个单独的计算所允许的最大时间为24小时,可任意设置。
vasp_path_dec = /opt/vasp.5.4.4/bin/vasp_gam # (path of VASP)
vasp_path_tsc = /opt/vasp.5.4.4/bin/vasp_std
# 对于TSC的计算,采用std版的VASP。对于DEC的单k点计算,采用gamma-only版的VASP。
job_name = submit_job # (name of script)
# 提交任务的脚本,命名为"submit_job",可任意设置。
potcar_path = /opt/POT/potpaw_PBE # (path of pseudopotentials)
# POTCAR的路径
max_job = 5
# 允许同时在跑的任务最大数
database_api = ******************* # (str-list type)
# 用于访问Materials Project数据库
level = 2 # (level=1: PBE+PBE; level=2: PBE+HSE; level=3: HSE+HSE)
# 对于超胞,使用PBE优化其原子位置,使用HSE计算其总能
min_atom = 96
max_atom = 96
# 我们希望生成的超胞大小为96个原子,且尽量使a=b=c,a⊥b⊥c
intrinsic = T # (default: T)
# 产生本征缺陷,V_Hf V_O Hf_O O_Hf Hf_i O_i
correction = FNV # (default: None)
# 带电缺陷的修正方案采用FNV修正
epsilon = 21.6
# HfO2的介电常数为21.6
Eg_real = 5.68 # (experimental band gap)
# HfO2的实验带隙值约为5.68 eV,DASP将根据此数据调整AEXX参数,从而使得无缺陷超胞的带隙值等于5.68 eV
ddc_temperature = 800 300
# 设置生长温度为800 K,工作温度为300 K
ddc_mass = 2.95 2.99
# 设置电子有效质量为2.95,空穴有效质量为2.99
5.2.1.2. 使用DASP产生必要输入文件¶
POSCAR 文件与 dasp.in 文件,执行 dasp 1 ,即可启动PREPARE模块,此后无需额外操作。DASP会输出 1prepare.out 文件记录程序的运行日志。5.2.1.3. PREPARE模块运行流程¶
产生超胞:
POSCAR_nearlycube :Cubic_cell
1.0
10.2770806222 0.0000000000 0.0000000000
0.0000000000 10.3884321420 0.0000000000
-1.7940733247 0.0000000000 10.5008134501
Hf O
32 64
Direct
0.3619599280 0.2715264169 0.1459735659
0.1380400719 0.0215264169 0.1040264340
0.1380400719 0.2284735830 0.3540264340
0.3619599280 0.4784735830 0.3959735659
...
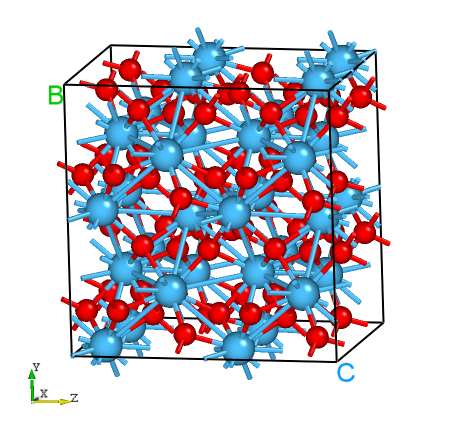
DASP产生的HfO2超胞的晶体结构。
马德隆常数计算:
1prepare.out 的输出如下:############ Prepare Files module start ############
Read the structure file POSCAR you provided
Get the refined cell POSCAR_refined from POSCAR
Generate the nearlycube cell POSCAR_nearlycube from POSCAR
Generate job script through dasp.in parameters
Generate single-point KPOINTS
Generate pseudopotential file POTCAR through potcar_dir you set
Generate commonly used vasp input file INCAR
Start the madelung constant calculation
Generate the madelung calculation directory
Generate madelung calculation POSCAR
Generate madelung calculation POTCAR
Generate madelung calculation INCAR
Generate madelung calculation KPOINTS
Generate madelung calculation job script
Job 103.host5 submitted: /home/fudan/HfO2/dec/madelung/static
Succeed job 103.host5: /home/fudan/HfO2/dec/madelung/static
The madelung constant calculation completed
The madelung constant = 2.841
HSE交换参数计算:
cd ./dec/AEXX
ls
0.25 AEXX.list
1prepare.out 可以看到如下日志:Start the HSE parameter AEXX calculation
Job 107.host5 submitted: /home/fudan/HfO2/dec/AEXX/0.25/static
Succeed job 107.host5: /home/fudan/HfO2/dec/AEXX/0.25/static
The HSE parameter AEXX calculation completed
The HSE parameter AEXX = 0.25
level = 2: Generate PBE relax vasp input file INCAR-relax
level = 2: Generate HSE static vasp input file INCAR-static
host超胞原子位置的优化:
POSCAR_final 。优化计算可见HfO2/dec/relax目录。同时也可以在 1prepare.out 可以DASP运行结束的标志,并告诉我们下一步需要做TSC模块的计算。Start the POSCAR_nearlycube relax calculation
Generate the POSCAR_nearlycube relax directory
Job 110.host5 submitted: /home/fudan/HfO2/dec/relax
Succeed job 110.host5: /home/fudan/HfO2/dec/relax
The POSCAR_nearlycube relax calculation completed
Get the final structure POSCAR_final
############ Prepare Files module end ############
DASP-PREPARE finished, please run DASP-TSC next
5.2.2. 热力学稳定性和元素化学势计算TSC¶
5.2.2.1. 运行TSC模块¶
1prepare.out 文件。等待程序执行完毕, 1prepare.out 有相应的完成标志。进入HfO2/dec目录。确认INCAR-relax,INCAR-static文件中的参数是可行的。(用户可修改INCAR,DASP将根据此目录中的INCAR做后续的计算)2tsc.out 。等待程序完成期间无需额外操作。5.2.2.2. TSC模块运行流程¶
host结构的总能计算(与MP参数保持一致):
Materials Project 数据库完全一致的输入参数(INCAR,KPOINTS,POTCAR)来对用户给定的原胞做结构优化和静态计算。因此,该计算得到的总能与MP数据库的总能是可比的。此步骤是为了得到影响HfO2稳定性的 关键杂相 。通过目录可以看到:cd tsc
cd HfO2/
ls
relaxation1 relaxation2 static
关键杂相判断:
2tsc.out 中可看到相关的信息:...
analysing the thermodynamic stability of HfO2.
key phases of HfO2 are: Hf O2 .
file key_phases_info_recalc.yaml generated.
analysing of HfO2 is done.
...
host与杂相结构的总能计算(PREPARE模块确定的参数):
2tsc.out 如下:...
Job 182.host5 submitted: /home/test/HfO2/tsc/HfO2/static_recalc
Job 183.host5 submitted: /home/test/HfO2/tsc/Hf/static_recalc
Job 184.host5 submitted: /home/test/HfO2/tsc/O2/static_recalc
Succeed job 182.host5: /home/test/HfO2/tsc/HfO2/static_recalc
Succeed job 183.host5: /home/test/HfO2/tsc/Hf/static_recalc
Succeed job 184.host5: /home/test/HfO2/tsc/O2/static_recalc
...
化学势的计算:
dasp.in :# 顺序与POSCAR中元素顺序一致,即第一列是Hf,第二列是O
E_pure = -11.1092 -8.2689
p1 = 0.0 -5.8748
p2 = -11.7496 0.0
2tsc.out 可以看到程序执行完毕的输出:dir '2d-figures','3d-figures','ori_data_MP' ready. try to read file: 'calc_list.yaml'.
analysing the thermodynamic stability of HfO2.
key phases of HfO2 are: Hf O2 .
analysing of HfO2 is done.
---------------------------
DASP-TSC finished
5.2.3. 缺陷形成能和转变能级计算DEC¶
5.2.3.1. 运行DEC模块¶
2tsc.out 文件。等待程序执行完毕, 2tsc.out 有相应的完成标志。打开HfO2/dasp.in,确认化学势已被程序自动输入。3dec.out 。等待程序完成期间无需额外操作。5.2.3.2. DEC模块运行流程¶
产生缺陷结构:
dasp.in 中的参数intrinsic = T,DEC模块将产生HfO2的本征缺陷,即生成HfO2/dec/Intrinsic_Defect计算目录,在其下面分别有空位缺陷V_Hf,V_O,反位缺陷Hf_O,O_Hf,间隙位缺陷Hf_i,O_i的缺陷结构和目录。根据对称性判断,HfO2晶格中不存在非等价的Hf原子,但存在两种不等价的O原子,因此V_O,Hf_O缺陷构型各有两种,V_Hf,O_Hf缺陷构型仅有一种,Hf_i,O_i的缺陷构型数量由用户输入参数决定。cd dec/Intrinsic_Defect/
ls
Hf_i Hf_O1 Hf_O2 host Intrinsic_Defect.list O_Hf1 O_i V_Hf1 V_O1 V_O2
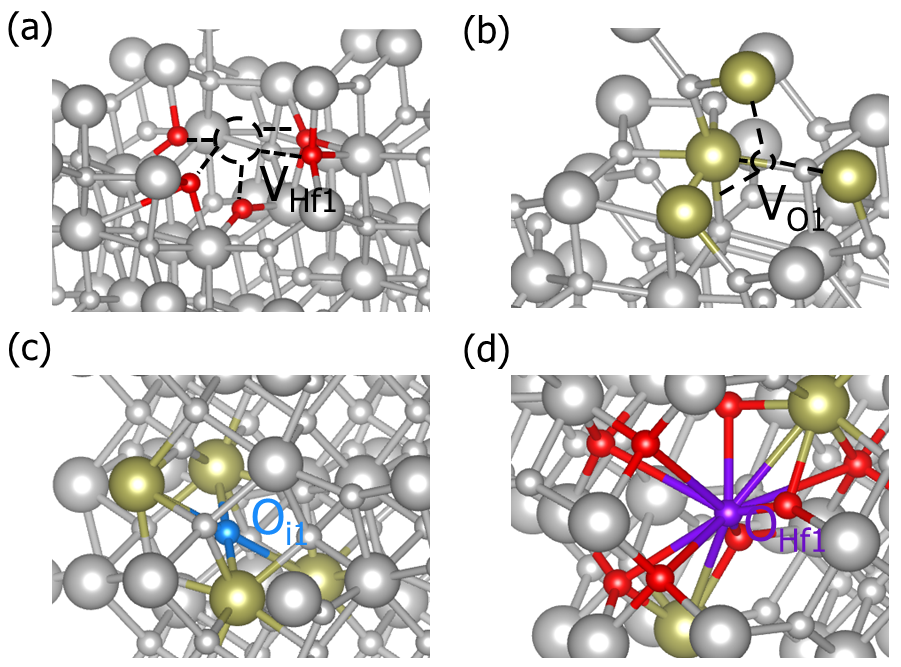
DASP产生的HfO2的部分缺陷结构。
3dec.out 看到DEC模块的输出如下:############ Neutral Defect module start ############
Make intrinsic defect directory Intrinsic_Defect
Generate host directory in Intrinsic_Defect
Start generating neutral vacancy defect
Generate neutral defect at: V_Hf1/initial_structure/q0
Generate neutral defect at: V_O1/initial_structure/q0
Generate neutral defect at: V_O2/initial_structure/q0
Neutral vacancy defect generation completed
Start generating neutral intrinsic antisite defect
Generate neutral defect at: O_Hf1/initial_structure/q0
Generate neutral defect at: Hf_O1/initial_structure/q0
Generate neutral defect at: Hf_O2/initial_structure/q0
Neutral intrinsic antisite defect generation completed
Start generating neutral intrinsic interstitial defect
Generate neutral defect at: Hf_i/random1/initial_structure/q0
Generate neutral defect at: Hf_i/random2/initial_structure/q0
Generate neutral defect at: Hf_i/random3/initial_structure/q0
Generate neutral defect at: Hf_i/random4/initial_structure/q0
Generate neutral defect at: Hf_i/random5/initial_structure/q0
Generate neutral defect at: Hf_i/random6/initial_structure/q0
Generate neutral defect at: O_i/random1/initial_structure/q0
Generate neutral defect at: O_i/random2/initial_structure/q0
Generate neutral defect at: O_i/random3/initial_structure/q0
Generate neutral defect at: O_i/random4/initial_structure/q0
Generate neutral defect at: O_i/random5/initial_structure/q0
Generate neutral defect at: O_i/random6/initial_structure/q0
Neutral intrinsic interstitial defect generation completed
############ Neutral Defect module end ############
提交各缺陷q=0计算任务:
dasp.in 中level = 2的参数),此步骤等待时间较长。可随时检查 3dec.out 文件。 3dec.out 中的相关信息如下所示:Job 198.host5 submitted: /data/HfO2/dec/Intrinsic_Defect/V_O2/initial_structure/q0
Job 200.host5 submitted: /data/HfO2/dec/Intrinsic_Defect/V_O1/initial_structure/q0
Job 202.host5 submitted: /data/HfO2/dec/Intrinsic_Defect/O_Hf1/initial_structure/q0
Job 204.host5 submitted: /data/HfO2/dec/Intrinsic_Defect/Hf_O1/initial_structure/q0
Job 206.host5 submitted: /data/HfO2/dec/Intrinsic_Defect/Hf_i/random5/initial_structure/q0
...
Succeed job 202.host5: /data/HfO2/dec/Intrinsic_Defect/O_Hf1/initial_structure/q0
Succeed job 198.host5: /data/HfO2/dec/Intrinsic_Defect/V_O2/initial_structure/q0
Failed job 204.host5: /data/HfO2/dec/Intrinsic_Defect/Hf_O1/initial_structure/q0
Succeed job 206.host5: /data/HfO2/dec/Intrinsic_Defect/Hf_i/random5/initial_structure/q0
...
产生带电缺陷的计算目录:
3dec.out 中的相关信息如下所示:############ Ionized Defect module start ############
Start generating ionized defects
Ionized defect path: /data/HfO2/dec/Intrinsic_Defect/Hf_O2/initial_structure/q+1
Ionized defect path: /data/HfO2/dec/Intrinsic_Defect/Hf_O2/initial_structure/q+2
Ionized defect path: /data/HfO2/dec/Intrinsic_Defect/Hf_O2/initial_structure/q+3
Ionized defect path: /data/HfO2/dec/Intrinsic_Defect/Hf_O2/initial_structure/q+4
Ionized defects generation completed
Start generating ionized defects
Ionized defect path: /data/HfO2/dec/Intrinsic_Defect/V_O2/initial_structure/q-2
Ionized defect path: /data/HfO2/dec/Intrinsic_Defect/V_O2/initial_structure/q-1
Ionized defect path: /data/HfO2/dec/Intrinsic_Defect/V_O2/initial_structure/q+1
Ionized defect path: /data/HfO2/dec/Intrinsic_Defect/V_O2/initial_structure/q+2
Ionized defects generation completed
Warning: static calculation undo in /data/HfO2/dec/Intrinsic_Defect/Hf_O1/initial_structure/q0/static, skipped generate ionized defect
The static calculation of /data/HfO2/dec/Intrinsic_Defect/Hf_i/random5/initial_structure/q0/static is skipped, skip ionized defect generation
...
提交各缺陷q≠0的计算任务:
dasp.in 中level = 2的参数),此步骤的等待时间比3.2.2的更长。 3dec.out 中的相关信息如下所示:############ AutoRun - Ionized Defect module start ############
Job 259.host5 submitted: /data/HfO2/dec/Intrinsic_Defect/Hf_O2/initial_structure/q+4
Job 261.host5 submitted: /data/HfO2/dec/Intrinsic_Defect/Hf_O2/initial_structure/q+1
Job 263.host5 submitted: /data/HfO2/dec/Intrinsic_Defect/Hf_O2/initial_structure/q+3
Job 265.host5 submitted: /data/HfO2/dec/Intrinsic_Defect/Hf_O2/initial_structure/q+2
Job 267.host5 submitted: /data/HfO2/dec/Intrinsic_Defect/V_O2/initial_structure/q+1
...
Succeed job 259.host5: /data/HfO2/dec/Intrinsic_Defect/Hf_O2/initial_structure/q+4
Succeed job 261.host5: /data/HfO2/dec/Intrinsic_Defect/Hf_O2/initial_structure/q+1
Succeed job 263.host5: /data/HfO2/dec/Intrinsic_Defect/Hf_O2/initial_structure/q+3
Succeed job 267.host5: /data/HfO2/dec/Intrinsic_Defect/V_O2/initial_structure/q+1
Succeed job 265.host5: /data/HfO2/dec/Intrinsic_Defect/Hf_O2/initial_structure/q+2
...
计算带电缺陷的修正:
dasp.in 中correction = FNV的参数),并计算其缺陷形成能和转变能级。由于之前计算错误(undo,failed,not converged)或者不进行后续计算(skip)的缺陷的报错信息,每个缺陷各价态的修正量和形成能的具体数值,都记录在 3dec.out 中:...
The formation energy (neutral) of Hf_O2 at p1 is 5.339603
The formation energy (neutral) of Hf_O2 at p2 is 22.964003
The FNV correction (q = 4) E_correct = 1.80575 eV
The transition level (0/4+) above VBM: 3.9438
The FNV correction (q = 1) E_correct = 0.159401 eV
The transition level (0/+) above VBM: 4.7144
The FNV correction (q = 3) E_correct = 1.02256 eV
The transition level (0/3+) above VBM: 4.2496
The FNV correction (q = 2) E_correct = 0.502853 eV
The transition level (0/2+) above VBM: 4.4441
...
The static calculation of /data/HfO2/dec/Intrinsic_Defect/O_i/random4/initial_structure/q0/static is skipped, skip formation energy calculation
Warning: calculation undo in /data/HfO2/dec/Intrinsic_Defect/Hf_O1/initial_structure/q0/static, skipped calculate formation energy
...
defect.log 文件中。输出形成能图像:
redo.in 的文件,在里面写入/home/test/HfO2/dec/Intrinsic_Defect/Hf_O1/initial_structure/q0。redo.in 重新计算该缺陷。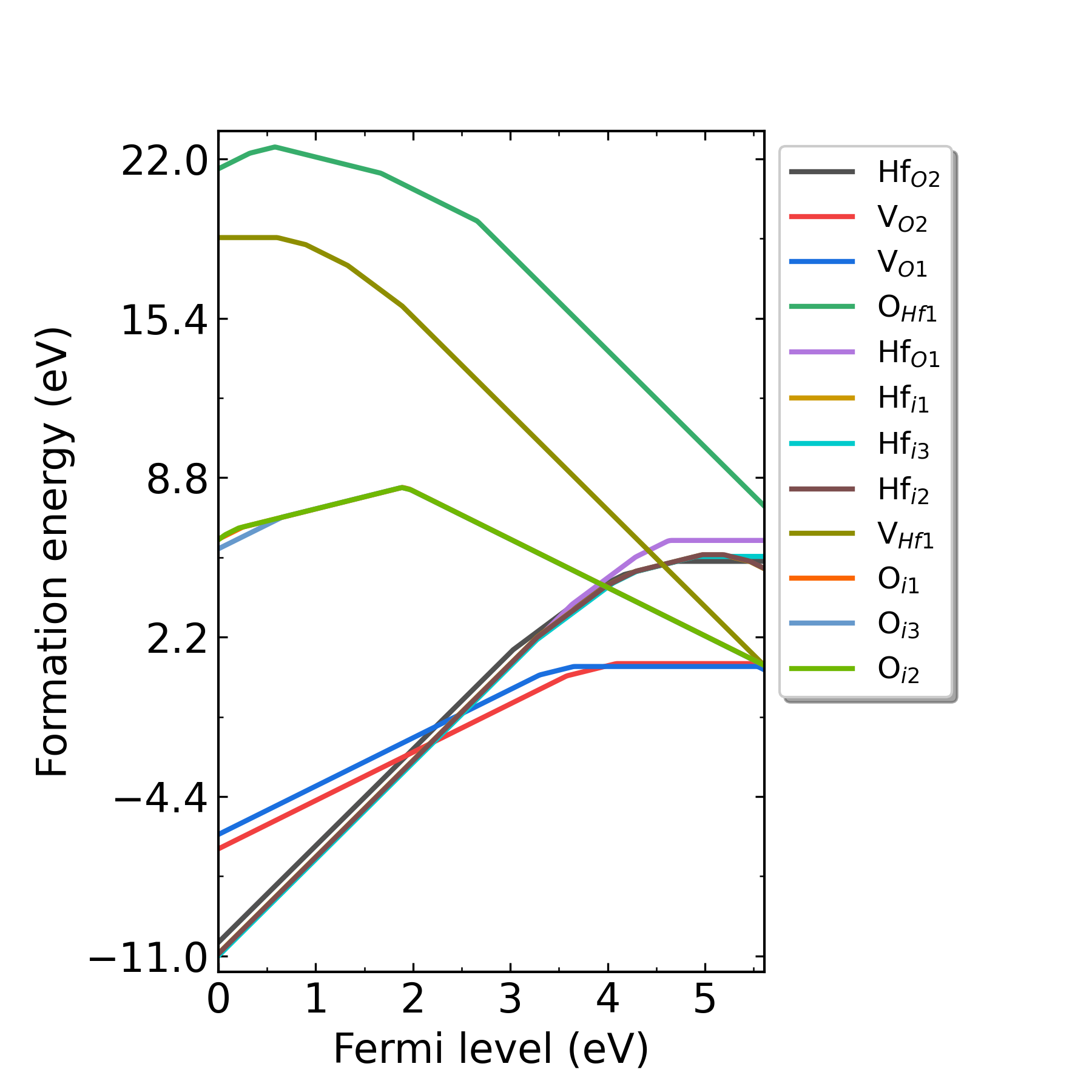
HfO2在p1处(Hf-rich)的缺陷形成能随费米能级的变化。
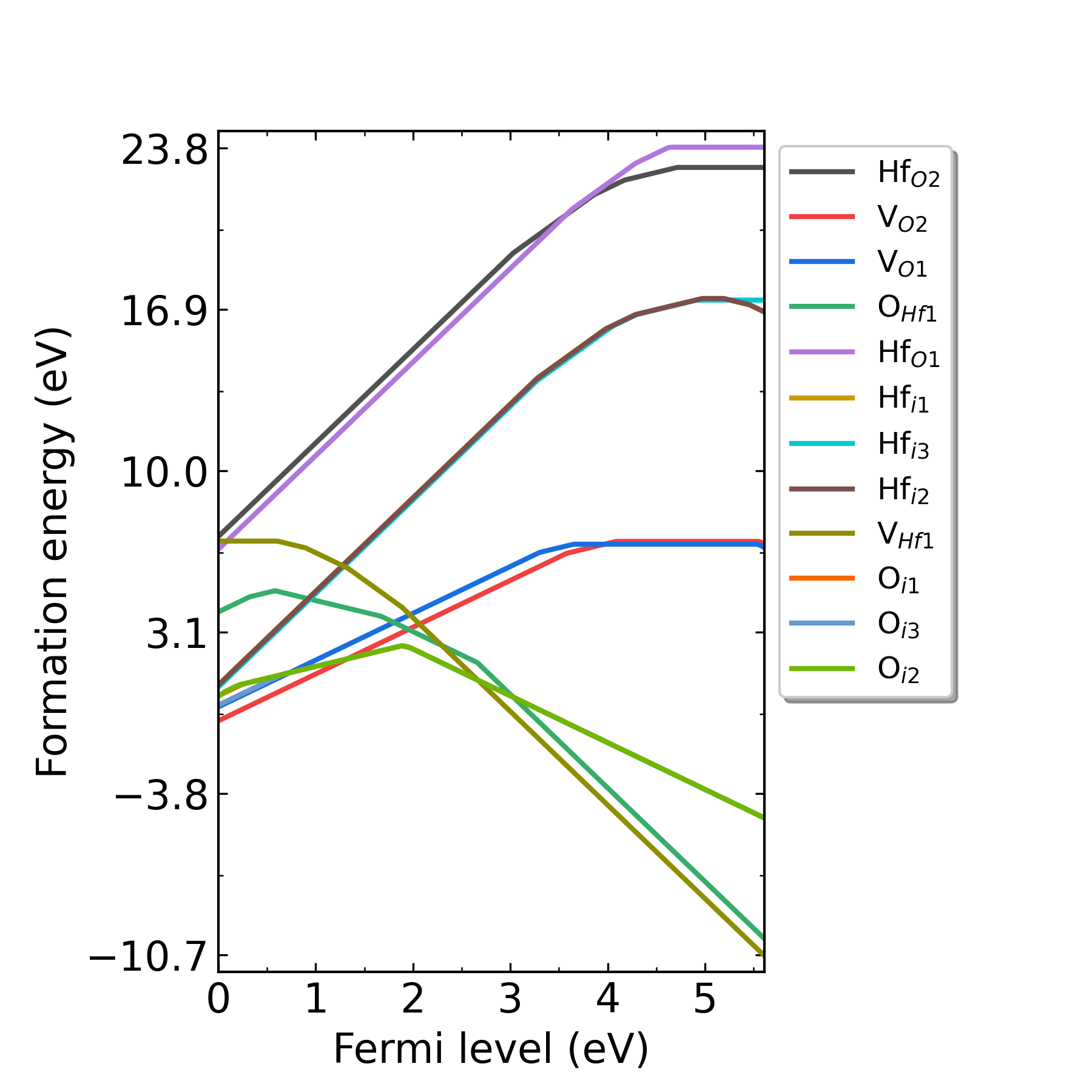
HfO2在p2处(O-rich)的缺陷形成能随费米能级的变化。
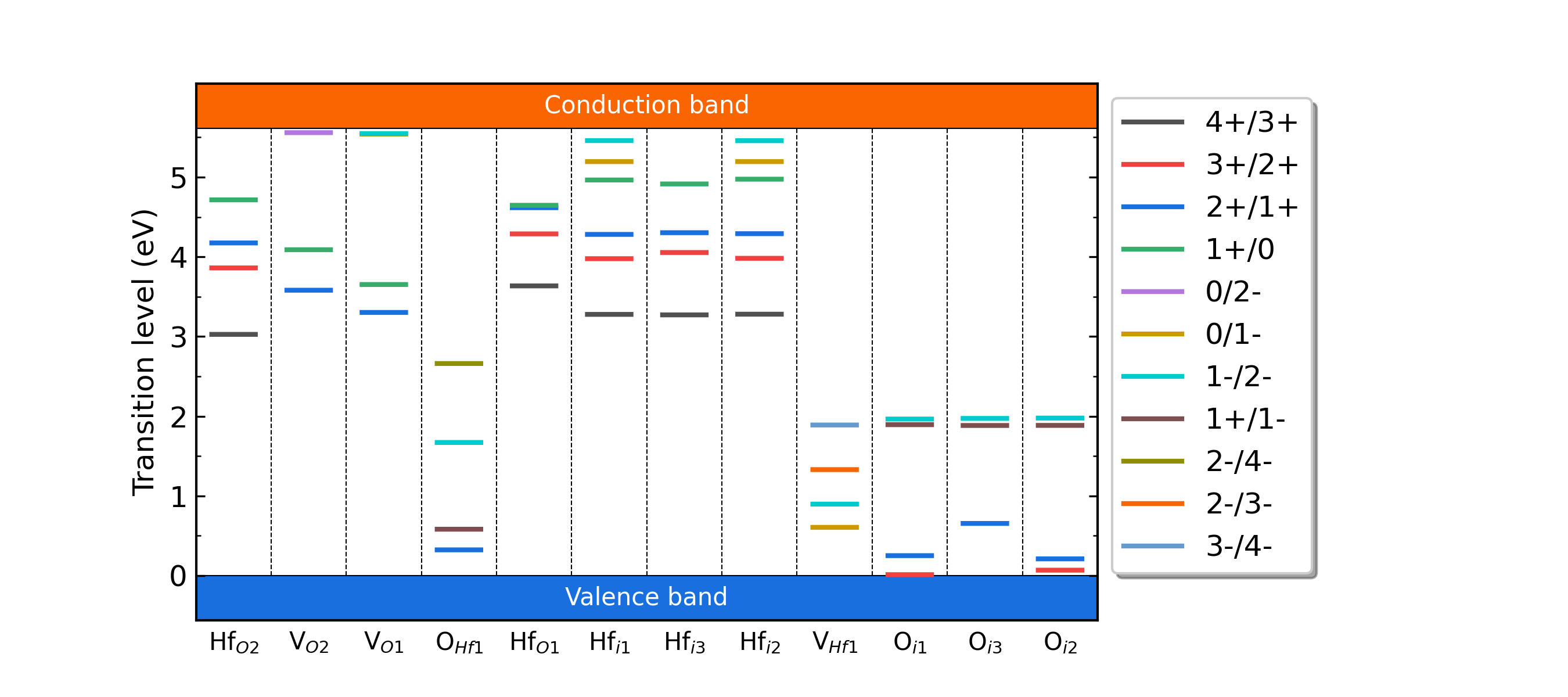
HfO2各缺陷的转变能级。
5.2.4. 缺陷浓度和费米能级计算DDC¶
5.2.4.1. 运行DDC模块¶
5.2.4.2. DDC模块运行流程¶
缺陷数据汇总:
DefectParams.txt 文件中。4ddc.out :############ Collecting information from DEC ############
Read defect types from DEC calculation successfully.
Defects considered in DDC calculation: ['Hf_O2', 'V_O2', 'V_O1', 'O_Hf1', 'Hf_O1', 'Hf_i-1', 'Hf_i-3', 'Hf_i-2', 'V_Hf1', 'O_i-1', 'O_i-3', 'O_i-2']
Chemical potentials change from p1 to p2.
Calculate gq for defect in each charge state.
Calculate Nsites for Hf_O2: 5.708701e+22 cm^-3.
Calculate Nsites for V_O2: 5.708701e+22 cm^-3.
Calculate Nsites for V_O1: 5.708701e+22 cm^-3.
Calculate Nsites for O_Hf1: 2.854350e+22 cm^-3.
Calculate Nsites for Hf_O1: 5.708701e+22 cm^-3.
Calculate Nsites for Hf_i-1: 2.854350e+22 cm^-3.
Calculate Nsites for Hf_i-3: 2.854350e+22 cm^-3.
Calculate Nsites for Hf_i-2: 2.854350e+22 cm^-3.
Calculate Nsites for V_Hf1: 2.854350e+22 cm^-3.
Calculate Nsites for O_i-1: 2.854350e+22 cm^-3.
Calculate Nsites for O_i-3: 2.854350e+22 cm^-3.
Calculate Nsites for O_i-2: 2.854350e+22 cm^-3.
############ Collecting information from DEC ############
DefectParams.txt 文件:800 300
2.950000 2.990000
5.611337
Hf_O2 5.708701e+22 1 4.714 2 4.444 1 4.25 2 3.944 1 x x x x x x x x 5.340000 22.964000
V_O2 5.708701e+22 1 4.089 2 3.835 1 x x x x 5.621 2 5.558 1 x x x x 1.106000 6.981000
V_O1 5.708701e+22 1 3.652 2 3.477 1 x x x x 5.539 2 5.542 1 x x x x 0.986000 6.861000
O_Hf1 2.854350e+22 1 0.79 2 0.557 1 x x x x 0.374 2 1.022 1 1.689 2 1.841 1 22.704000 5.080000
Hf_O1 5.708701e+22 1 4.646 2 4.631 1 4.517 2 4.296 1 x x x x x x x x 6.205000 23.830000
Hf_i-1 2.854350e+22 1 4.964 2 4.622 1 4.407 2 4.125 1 5.194 2 5.326 1 x x x x 5.601000 17.351000
Hf_i-3 2.854350e+22 1 4.914 2 4.608 1 4.423 2 4.135 1 x x x x x x x x 5.544000 17.294000
Hf_i-2 2.854350e+22 1 4.974 2 4.632 1 4.415 2 4.131 1 5.194 2 5.326 1 x x x x 5.614000 17.364000
V_Hf1 2.854350e+22 1 x x x x x x x x 0.606 2 0.751 1 0.944 2 1.18 1 18.743000 6.994000
O_i-1 2.854350e+22 1 2.09 2 1.17 1 0.784 2 x x 1.697 2 1.831 1 x x x x 8.596000 2.722000
O_i-3 2.854350e+22 1 2.093 2 1.374 1 0.814 2 x x 1.675 2 1.824 1 x x x x 8.606000 2.731000
O_i-2 2.854350e+22 1 2.095 2 1.153 1 0.791 2 x x 1.674 2 1.825 1 x x x x 8.606000 2.731000
生长温度下自洽计算:
DDC模块在T=800 K的时候计算其缺陷浓度和载流子浓度,并根据电中性条件自洽求解费米能级。
工作温度下自洽计算:
DDC模块在T=300 K的时候重新分布每个缺陷各价态的浓度,并根据电中性条件再次自洽求解费米能级。
输出缺陷浓度:
DDC模块在HfO2/ddc目录下,输出三个文件:费米能级文件 Fermi.dat ,载流子浓度文件 Carrier.dat ,缺陷浓度文件 Defect_charge.dat 。可使用Origin画图。
此外,DDC模块会自动根据三个文件画图,产生density.png文件。如下图所示:
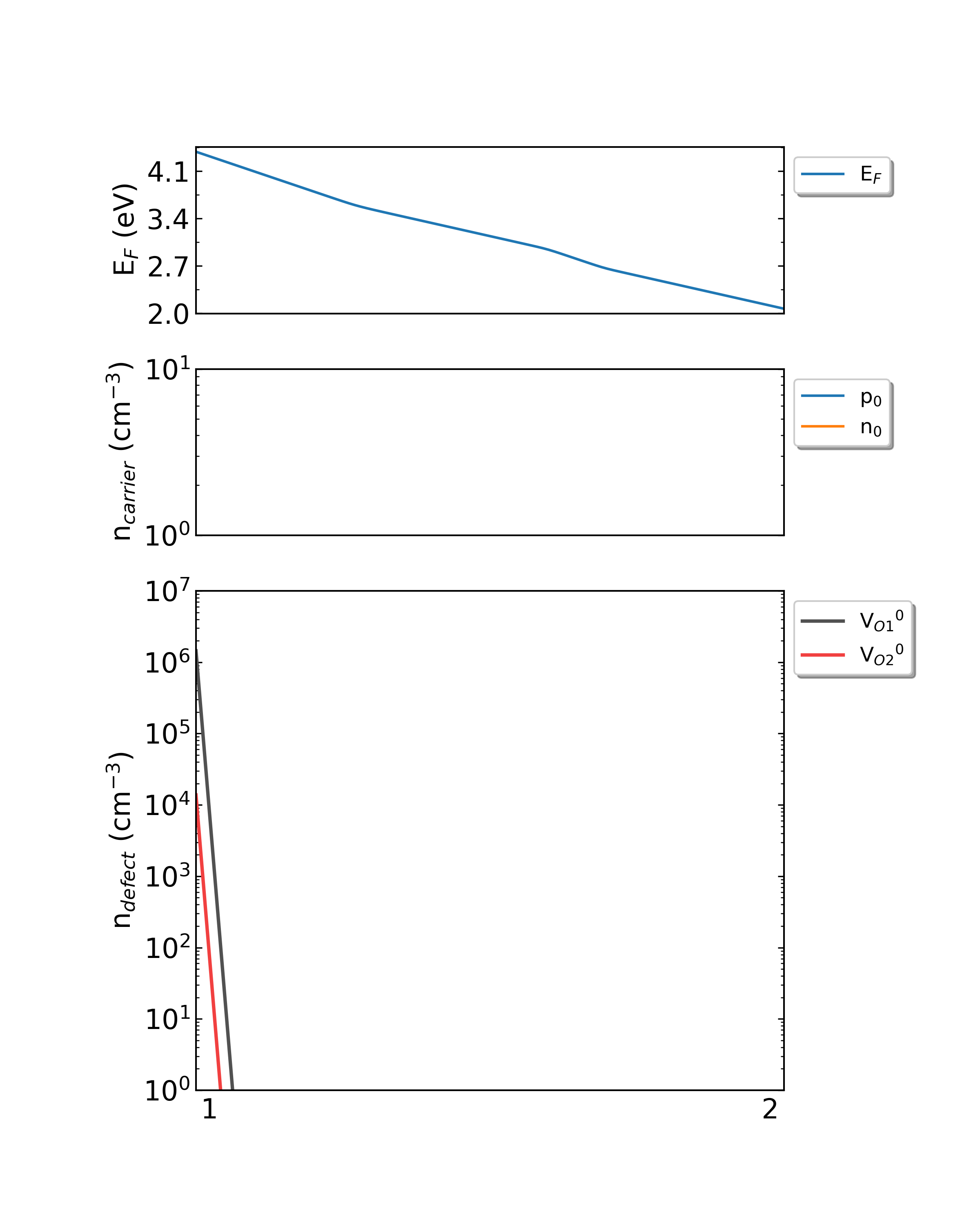
生长温度为300K时,HfO2从p1(Hf-rich)到p2(O-rich)的费米能级、载流子浓度、缺陷浓度。
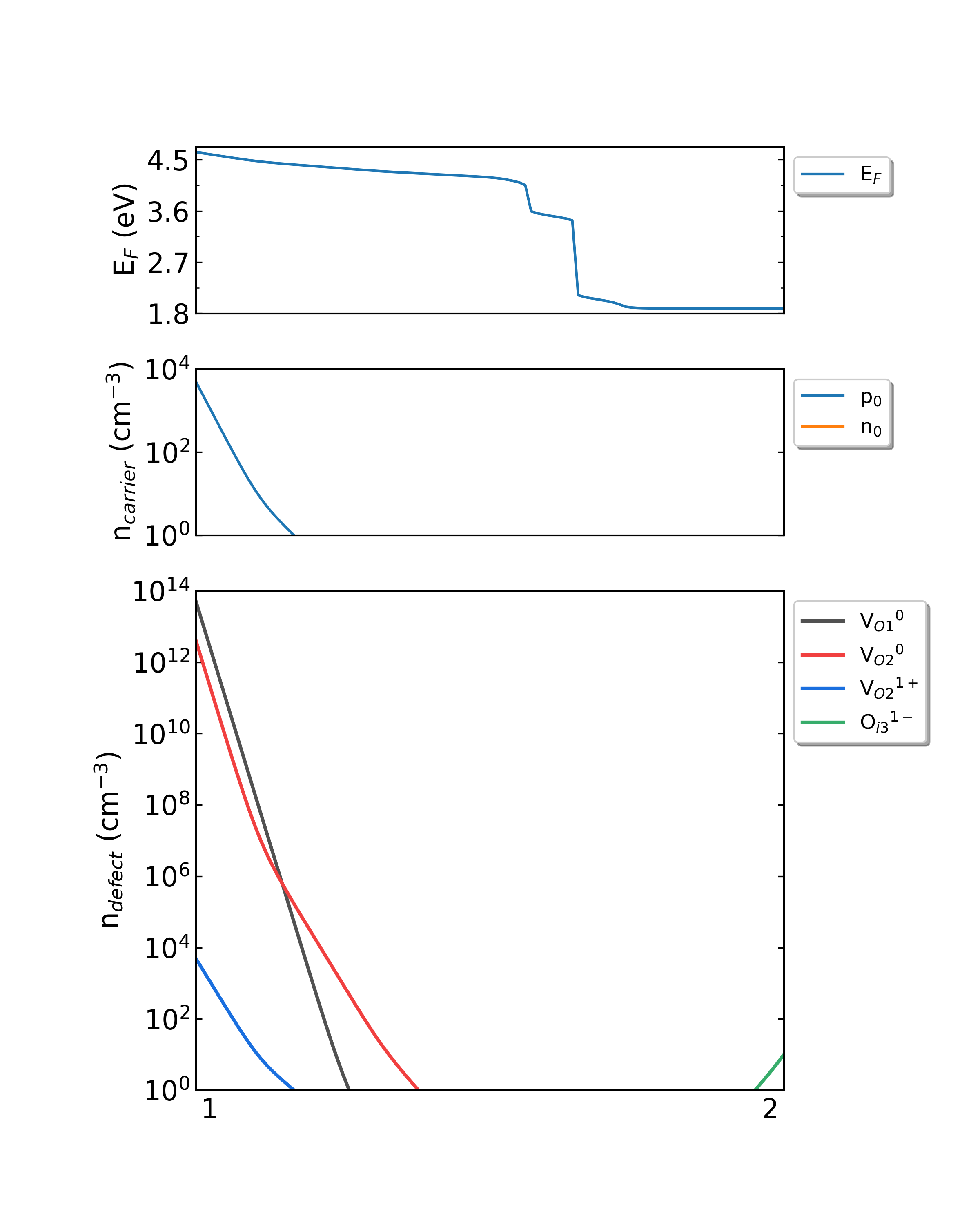
生长温度为550K时,HfO2从p1(Hf-rich)到p2(O-rich)的费米能级、载流子浓度、缺陷浓度。
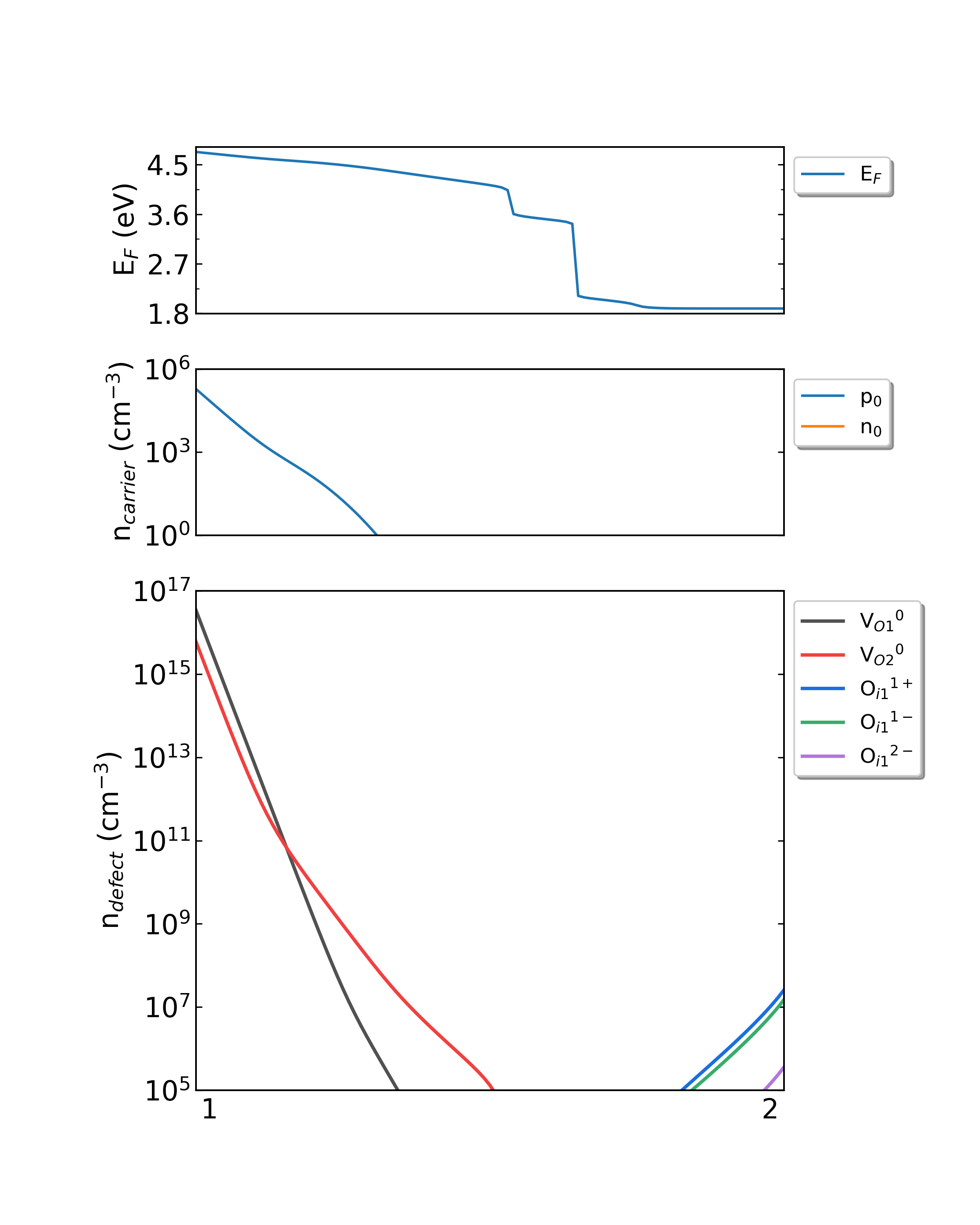
生长温度为800K时,HfO2从p1(Hf-rich)到p2(O-rich)的费米能级、载流子浓度、缺陷浓度。
5.3. H掺杂Ga2O3的缺陷计算¶
5.3.1. 准备计算PREPARE¶
5.3.1.1. 准备POSCAR与dasp.in¶
Materials Project 数据库中找到Ga2O3的 POSCAR 文件,显示如下:Ga8 O12
1.0
12.2299995422 0.0000000000 0.0000000000
0.0000000000 3.0399999619 0.0000000000
-1.3736609922 0.0000000000 5.6349851545
Ga O
8 12
Direct
0.158409998 0.500000000 0.314081997
0.341589987 0.000000000 0.685917974
0.089878000 0.000000000 0.794761002
0.410122007 0.500000000 0.205238998
0.658410013 0.000000000 0.314081997
0.841589987 0.500000000 0.685917974
0.589878023 0.500000000 0.794761002
0.910121977 0.000000000 0.205238998
0.495896995 0.000000000 0.256491005
0.004103000 0.500000000 0.743508995
0.173598006 0.000000000 0.564293981
0.326402009 0.500000000 0.435705990
0.336497009 0.500000000 0.891047001
0.163503006 0.000000000 0.108952999
0.995896995 0.500000000 0.256491005
0.504103005 0.000000000 0.743508995
0.673597991 0.500000000 0.564293981
0.826402009 0.000000000 0.435705990
0.836497009 0.000000000 0.891047001
0.663502991 0.500000000 0.108952999
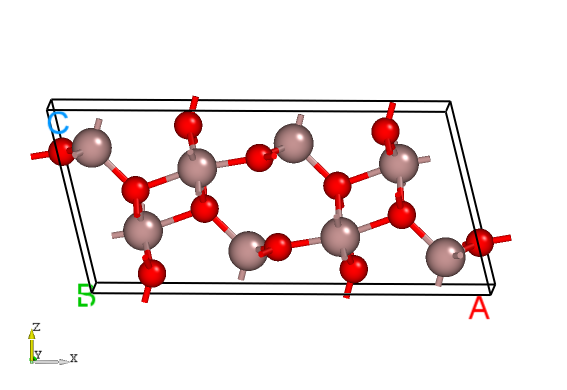
Ga2O3的晶体结构。
############## Job Scheduling ##############
cluster = SLURM # (job scheduling system)
node_number = 4 # (number of node)
core_per_node = 52 # (core per node)
queue = batch # (name of queue/partition)
max_time = 24:00:00 # (maximum time for a single DFT calculation)
vasp_path_dec = /opt/vasp.5.4.4/bin/vasp_gam # (path of VASP)
vasp_path_tsc = /opt/vasp.5.4.4/bin/vasp_std
job_name = submit_job # (name of script)
potcar_path = /opt/POT/potpaw_PBE # (path of pseudopotentials)
max_job = 5
############## TSC Module ##############
database_api = ******************* # (str-list type)
############## DEC Module ##############
level = 2 # (level=1: PBE+PBE; level=2: PBE+HSE; level=3: HSE+HSE)
min_atom = 200
max_atom = 250
intrinsic = F # (default: T)
doping = T # (default: F)
impurity = H
correction = FNV # (default: none)
epsilon = 10.8
Eg_real = 4.9 # (experimental band gap)
############## DDC Module ##############
ddc_temperature = 1000 300
ddc_mass = 0.23 2.90
dasp.in 中所有列出的参数进行说明。cluster = SLURM
# 表示使用集群的队列系统为SLURM
node_number = 4
# 对于每个单独的计算,使用4个节点
core_per_node = 52
# 对于每个节点,使用52个核。因此对于每个单独的计算,总共使用4*52=208核
queue = batch
# 使用名为"batch"的队列进行计算。因此,在设置dasp.in参数之前,需要确认超算/集群上的队列名、节点、核数
max_time = 24:00:00 # (maximum time for a single DFT calculation)
# 每个单独的计算所允许的最大时间为24小时,可任意设置。
vasp_path_dec = /opt/vasp.5.4.4/bin/vasp_gam # (path of VASP)
vasp_path_tsc = /opt/vasp.5.4.4/bin/vasp_std
# 对于TSC的计算,采用std版的VASP。对于DEC的单k点计算,采用gam版的VASP。
job_name = submit_job # (name of script)
# 提交任务的脚本,命名为"submit_job",可任意设置。
potcar_path = /opt/POT/potpaw_PBE # (path of pseudopotentials)
# POTCAR的路径
max_job = 5
# 允许同时在跑的任务最大数
database_api = ******************* # (str-list type)
# 用于访问Materials Project数据库
level = 2 # (level=1: PBE+PBE; level=2: PBE+HSE; level=3: HSE+HSE)
# 对于超胞,使用PBE优化其原子位置,使用HSE计算其总能
min_atom = 200
max_atom = 250
# 我们希望生成的超胞的原子数为200-250个,且尽量使a=b=c,a⊥b⊥c
intrinsic = F # (default: T)
# 不产生本征缺陷
doping = T # (default: F)
# 产生掺杂缺陷
impurity = H
# 掺杂H元素,产生掺杂缺陷H_Ga H_O H_i
correction = FNV # (default: none)
# 带电缺陷的修正方案采用FNV修正
epsilon = 10.8
# Ga2O3的介电常数为10.8
Eg_real = 4.9 # (experimental band gap)
# Ga2O3的实验带隙值约为4.9 eV,DASP将根据此数据调整AEXX参数,从而使得无缺陷超胞的带隙值等于4.9 eV
ddc_temperature = 1000 300
# 设置生长温度为1000 K,工作温度为300 K
ddc_mass = 0.23 4.21
# 设置电子有效质量为0.23,空穴有效质量为4.21
5.3.1.2. 使用DASP产生必要输入文件¶
POSCAR 文件与 dasp.in 文件,执行 dasp 1 ,即可启动PREPARE模块,此后无需额外操作。DASP会输出 1prepare.out 文件记录程序的运行日志。5.3.1.2. PREPARE模块运行流程¶
产生超胞:
POSCAR_nearlycube :Cubic_cell
1.0
18.9887859181 0.0000000000 0.0000000000
-1.4600617135 9.0023673390 0.0000000000
0.7906182108 0.1282275357 14.7068652328
Ga O
96 144
Direct
0.1256845017 0.0418948339 0.5327255075
0.3743154982 0.2914384994 0.4672744924
0.2212487535 0.2404162511 0.3686292617
0.2787512464 0.0929170821 0.6313707382
0.8756845017 0.1252281672 0.2827255075
0.1243154982 0.0414384994 0.2172744924
...
DASP产生的Ga2O3超胞的晶体结构。
马德隆常数计算:
1prepare.out 的输出如下:############ Prepare Files module start ############
Read the structure file POSCAR you provided
Get the refined cell POSCAR_refined from POSCAR
Generate the nearlycube cell POSCAR_nearlycube from POSCAR
Generate job script through dasp.in parameters
Generate single-point KPOINTS
Generate pseudopotential file POTCAR through potcar_dir you set
Generate commonly used vasp input file INCAR
Start the madelung constant calculation
Generate the madelung calculation directory
Generate madelung calculation POSCAR
Generate madelung calculation POTCAR
Generate madelung calculation INCAR
Generate madelung calculation KPOINTS
Generate madelung calculation job script
Job 503.host5 submitted: /data2/home/chensy/zzn/doping-Ga2O3/dec/madelung/static
Succeed job 503.host5: /data2/home/chensy/zzn/doping-Ga2O3/dec/madelung/static
The madelung constant calculation completed
The madelung constant = 2.411
HSE交换参数计算:
cd ./dec/AEXX
ls
0.25 0.3 0.3292780889291405 AEXX.list
1prepare.out 可以看到如下日志:Start the HSE parameter AEXX calculation
Job 507.host5 submitted: /data2/home/chensy/zzn/doping-Ga2O3/dec/AEXX/0.25/static
Succeed job 507.host5: /data2/home/chensy/zzn/doping-Ga2O3/dec/AEXX/0.25/static
Job 508.host5 submitted: /data2/home/chensy/zzn/doping-Ga2O3/dec/AEXX/0.3/static
Succeed job 508.host5: /data2/home/chensy/zzn/doping-Ga2O3/dec/AEXX/0.3/static
Job 509.host5 submitted: /data2/home/chensy/zzn/doping-Ga2O3/dec/AEXX/0.3292780889291405/static
Succeed job 509.host5: /data2/home/chensy/zzn/doping-Ga2O3/dec/AEXX/0.3292780889291405/static
The HSE parameter AEXX calculation completed
The HSE parameter AEXX = 0.33
level = 2: Generate PBE relax vasp input file INCAR-relax
level = 2: Generate HSE static vasp input file INCAR-static
host超胞原子位置的优化:
POSCAR_final 。优化计算可见doping-Ga2O3/dec/relax目录。同时也可以在 1prepare.out 可以DASP运行结束的标志,并告诉我们下一步需要做TSC模块的计算。Start the POSCAR_nearlycube relax calculation
Generate the POSCAR_nearlycube relax directory
Job 510.host5 submitted: /data2/home/chensy/zzn/doping-Ga2O3/dec/relax
Succeed job 510.host5: /data2/home/chensy/zzn/doping-Ga2O3/dec/relax
The POSCAR_nearlycube relax calculation completed
Get the final structure POSCAR_final
############ Prepare Files module end ############
DASP-PREPARE finished, please run DASP-TSC next
5.3.2. 热力学稳定性和元素化学势计算TSC¶
5.3.2.1. 运行TSC模块¶
1prepare.out 文件。等待程序执行完毕, 1prepare.out 有相应的完成标志。进入doping-Ga2O3/dec目录。确认INCAR-relax,INCAR-static文件中的参数是可行的。(用户可修改INCAR,DASP将根据此目录中的INCAR做后续的计算)2tsc.out 。等待程序完成期间无需额外操作。5.3.2.2. TSC模块运行流程¶
host结构的总能计算(与MP参数保持一致):
Materials Project 数据库完全一致的输入参数(INCAR,KPOINTS,POTCAR)来对用户给定的原胞做结构优化和静态计算。因此,该计算得到的总能与MP数据库的总能是可比的。此步骤是为了得到影响Ga2O3稳定性的 关键杂相 。通过目录可以看到:cd tsc
cd Ga2O3/
ls
relaxation1 relaxation2 static
关键杂相判断:
2tsc.out 中可看到相关的信息:...
analysing the thermodynamic stability of Ga2O3.
The stability of Ga2O3 is: True.
key phases of Ga2O3 are: Ga O2 .
key phases of H doped Ga2O3 are: H2 GaHO2 .
analysing of Ga2O3 is done.
sub-module of tsc: 'auto thermodynamic calculation' ends successfully.
...
host与杂相结构的总能计算(PREPARE模块确定的参数):
2tsc.out 如下:...
Job 520.host5 submitted: /data2/home/chensy/zzn/doping-Ga2O3/tsc/GaHO2/static_recalc
Job 521.host5 submitted: /data2/home/chensy/zzn/doping-Ga2O3/tsc/H2/static_recalc
Succeed job 520.host5: /data2/home/chensy/zzn/doping-Ga2O3/tsc/GaHO2/static_recalc
Succeed job 521.host5: /data2/home/chensy/zzn/doping-Ga2O3/tsc/H2/static_recalc
...
化学势的计算:
dasp.in :# 顺序与POSCAR中元素顺序一致,即第一列是Ga,第二列是O,第三列是掺杂元素H
E_pure = -4.1294 -9.4157 -4.0091
p1 = 0.0 -3.72 0.0
p2 = -5.5801 0.0 -1.7472
2tsc.out 可以看到程序执行完毕的输出:dir '2d-figures','3d-figures','ori_data_MP' ready. analysing the thermodynamic stability of Ga2O3.
The stability of Ga2O3 is: True.
key phases of Ga2O3 are: Ga O2 .
key phases of H doped Ga2O3 are: H2 GaHO2 .
analysing of Ga2O3 is done.
sub-module of tsc: 'auto thermodynamic calculation' ends successfully.
--------------------------
DASP-TSC finished
5.3.3. 缺陷形成能和转变能级计算DEC¶
5.3.3.1. 运行DEC模块¶
2tsc.out 文件。等待程序执行完毕, 2tsc.out 有相应的完成标志。打开doping-Ga2O3/dasp.in,确认化学势已被程序自动输入。3dec.out 。等待程序完成期间无需额外操作。5.3.3.2. DEC模块运行流程¶
产生缺陷结构:
dasp.in 中的参数doping = T和impurity = H,DEC模块将产生Ga2O3的掺杂H的缺陷,即生成doping-Ga2O3/dec/Doping_H计算目录,在其下面分别有替位缺陷H_Ga,H_O,间隙位缺陷H_i的缺陷结构和目录。根据对称性判断,Ga2O3晶格中存在两种不等价的Ga原子,但存在三种不等价的O原子,因此H_Ga缺陷构型有两种,H_O缺陷构型有三种,H_i的缺陷构型数量由用户输入参数决定。cd dec/Doping_H/
ls
Doping_H.list H_Ga1 H_Ga2 H_i H_O1 H_O2 H_O3 host
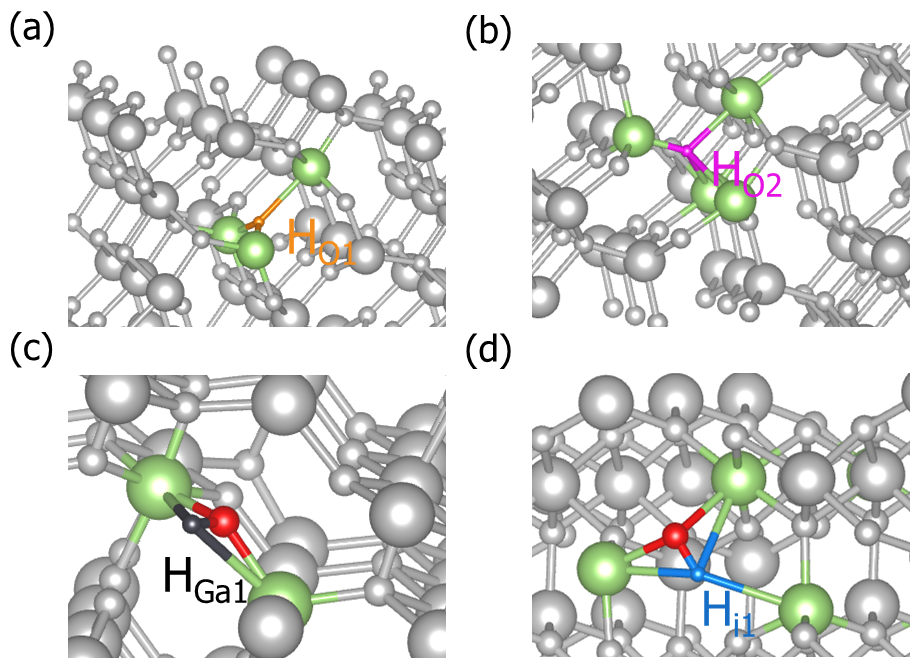
DASP产生的掺杂H的Ga2O3的部分缺陷结构。
3dec.out 看到DEC模块的输出如下:############ Neutral Defect module start ############
Make doping defect directory Doping_H
Generate host directory in Doping_H
Start generating neutral doping_H antisite defect
Generate neutral defect at: H_Ga1/initial_structure/q0
Generate neutral defect at: H_Ga2/initial_structure/q0
Generate neutral defect at: H_O1/initial_structure/q0
Generate neutral defect at: H_O2/initial_structure/q0
Generate neutral defect at: H_O3/initial_structure/q0
Neutral doping_H substitution defect generation completed
Start generating neutral doping_H interstitial defect
Generate neutral defect at: H_i/random1/initial_structure/q0
Generate neutral defect at: H_i/random2/initial_structure/q0
Generate neutral defect at: H_i/random3/initial_structure/q0
Generate neutral defect at: H_i/random4/initial_structure/q0
Generate neutral defect at: H_i/random5/initial_structure/q0
Generate neutral defect at: H_i/random6/initial_structure/q0
Neutral doping_H interstitial defect generation completed
############ Neutral Defect module end ############
提交各缺陷q=0计算任务:
dasp.in 中level = 2的参数),此步骤等待时间较长。可随时检查 3dec.out 文件。 3dec.out 中的相关信息如下所示:Job 598.host5 submitted: /data2/home/chensy/zzn/doping-Ga2O3/H_Ga1/initial_structure/q0
Job 600.host5 submitted: /data2/home/chensy/zzn/doping-Ga2O3/H_Ga2/initial_structure/q0
Job 602.host5 submitted: /data2/home/chensy/zzn/doping-Ga2O3/H_O1/initial_structure/q0
Job 604.host5 submitted: /data2/home/chensy/zzn/doping-Ga2O3/H_O2/initial_structure/q0
Job 606.host5 submitted: /data2/home/chensy/zzn/doping-Ga2O3/H_O3/initial_structure/q0
...
Succeed job 602.host5: /data2/home/chensy/zzn/doping-Ga2O3/H_O1/initial_structure/q0
Succeed job 600.host5: /data2/home/chensy/zzn/doping-Ga2O3/H_Ga2/initial_structure/q0
Succeed job 598.host5: /data2/home/chensy/zzn/doping-Ga2O3/H_Ga1/initial_structure/q0
Succeed job 604.host5: /data2/home/chensy/zzn/doping-Ga2O3/H_O2/initial_structure/q0
Succeed job 606.host5: /data2/home/chensy/zzn/doping-Ga2O3/H_O3/initial_structure/q0
...
产生带电缺陷的计算目录:
3dec.out 中的相关信息如下所示:############ Ionized Defect module start ############
Start generating ionized defects
Ionized defect path: /data2/home/chensy/zzn/doping-Ga2O3/H_O1/initial_structure/q+1
Ionized defect path: /data2/home/chensy/zzn/doping-Ga2O3/H_O1/initial_structure/q+2
Ionized defect path: /data2/home/chensy/zzn/doping-Ga2O3/H_O1/initial_structure/q+3
Ionized defect path: /data2/home/chensy/zzn/doping-Ga2O3/H_O1/initial_structure/q+4
Ionized defects generation completed
Start generating ionized defects
Ionized defect path: /data2/home/chensy/zzn/doping-Ga2O3/H_O2/initial_structure/q+1
Ionized defect path: /data2/home/chensy/zzn/doping-Ga2O3/H_O2/initial_structure/q+2
Ionized defect path: /data2/home/chensy/zzn/doping-Ga2O3/H_O2/initial_structure/q+3
Ionized defect path: /data2/home/chensy/zzn/doping-Ga2O3/H_O2/initial_structure/q+4
Ionized defects generation completed
The static calculation of /data2/home/chensy/zzn/doping-Ga2O3/H_i/random3/initial_structure/q0/static is skipped, skip ionized defect generation
...
提交各缺陷q≠0的计算任务:
dasp.in 中level = 2的参数),此步骤的等待时间比3.2.2的更长。 3dec.out 中的相关信息如下所示:############ AutoRun - Ionized Defect module start ############
Job 659.host5 submitted: /data2/home/chensy/zzn/doping-Ga2O3/H_O1/initial_structure/q+2
Job 661.host5 submitted: /data2/home/chensy/zzn/doping-Ga2O3/H_O1/initial_structure/q+1
Job 663.host5 submitted: /data2/home/chensy/zzn/doping-Ga2O3/H_O1/initial_structure/q+3
Job 665.host5 submitted: /data2/home/chensy/zzn/doping-Ga2O3/H_O1/initial_structure/q-1
Job 667.host5 submitted: /data2/home/chensy/zzn/doping-Ga2O3/H_O2/initial_structure/q+2
...
Succeed job 659.host5: /data2/home/chensy/zzn/doping-Ga2O3/H_O1/initial_structure/q+2
Succeed job 661.host5: /data2/home/chensy/zzn/doping-Ga2O3/H_O1/initial_structure/q+1
Succeed job 663.host5: /data2/home/chensy/zzn/doping-Ga2O3/H_O1/initial_structure/q+3
Succeed job 665.host5: /data2/home/chensy/zzn/doping-Ga2O3/H_O1/initial_structure/q-1
Succeed job 667.host5: /data2/home/chensy/zzn/doping-Ga2O3/H_O2/initial_structure/q+2
...
计算带电缺陷的修正:
dasp.in 中correction = FNV的参数),并计算其缺陷形成能和转变能级。由于之前计算错误(undo,failed,not converged)或者不进行后续计算(skip)的缺陷的报错信息,每个缺陷各价态的修正量和形成能的具体数值,都记录在 3dec.out 中:...
The formation energy (neutral) of H_O1 at p1 is 1.84 eV
The formation energy (neutral) of H_O1 at p2 is 7.42 eV
The FNV correction (q = 2) E_correct = -0.247 eV
The transition level (0/2+) above VBM: 2.627 eV
The FNV correction (q = 1) E_correct = 0.082 eV
The transition level (0/+) above VBM: 4.818 eV
The FNV correction (q = 3) E_correct = -0.075 eV
The transition level (0/3+) above VBM: 1.739 eV
The FNV correction (q = -1) E_correct = -0.056 eV
The transition level (-/0) above VBM: 4.769 eV
...
The static calculation of /data2/home/chensy/zzn/doping-Ga2O3/dec/Doping_H/H_i/random3/initial_structure/q0/static is skipped, skip formation energy calculation
...
defect.log 文件中。输出形成能图像:
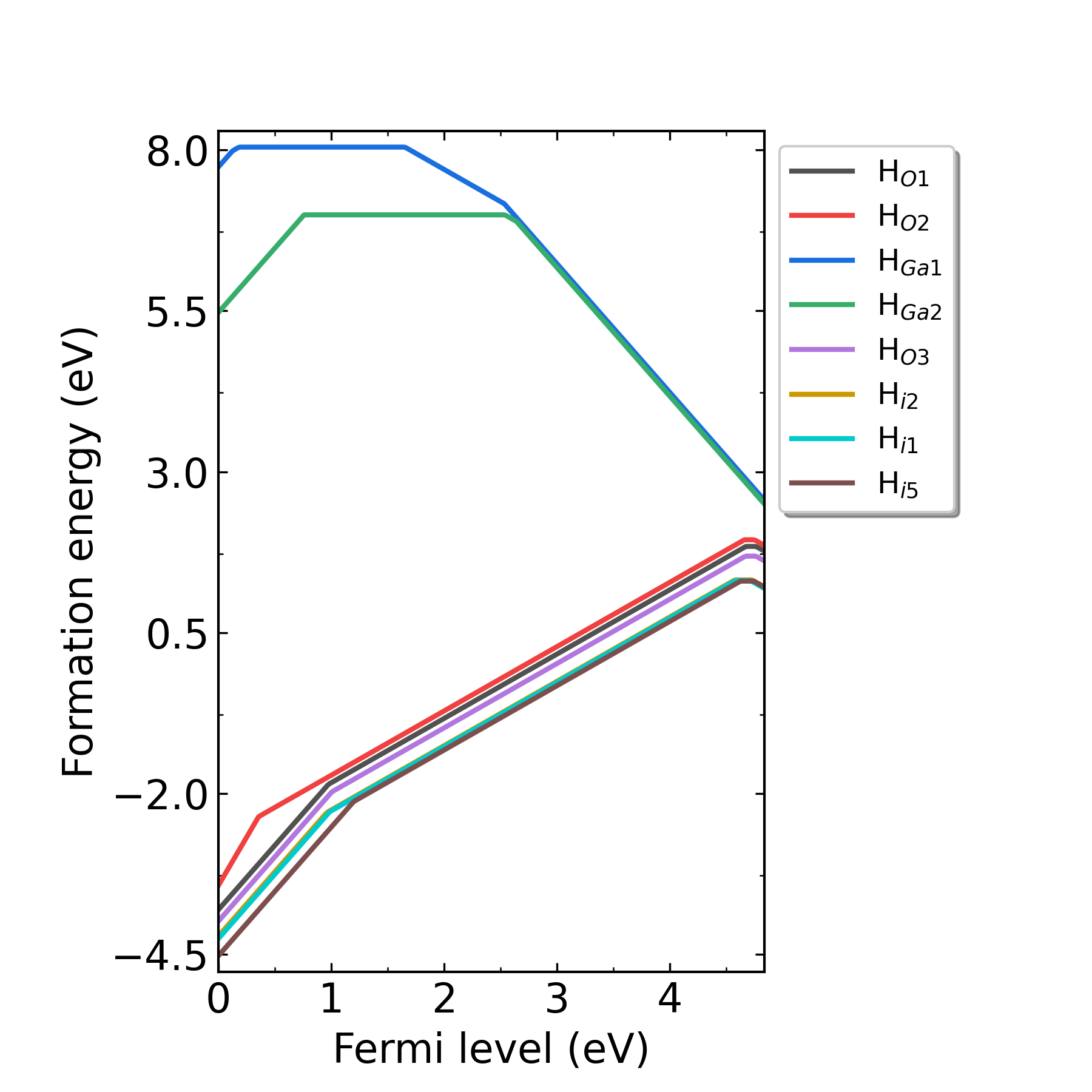
掺杂H的Ga2O3在p1处(Ga-rich)的缺陷形成能随费米能级的变化。
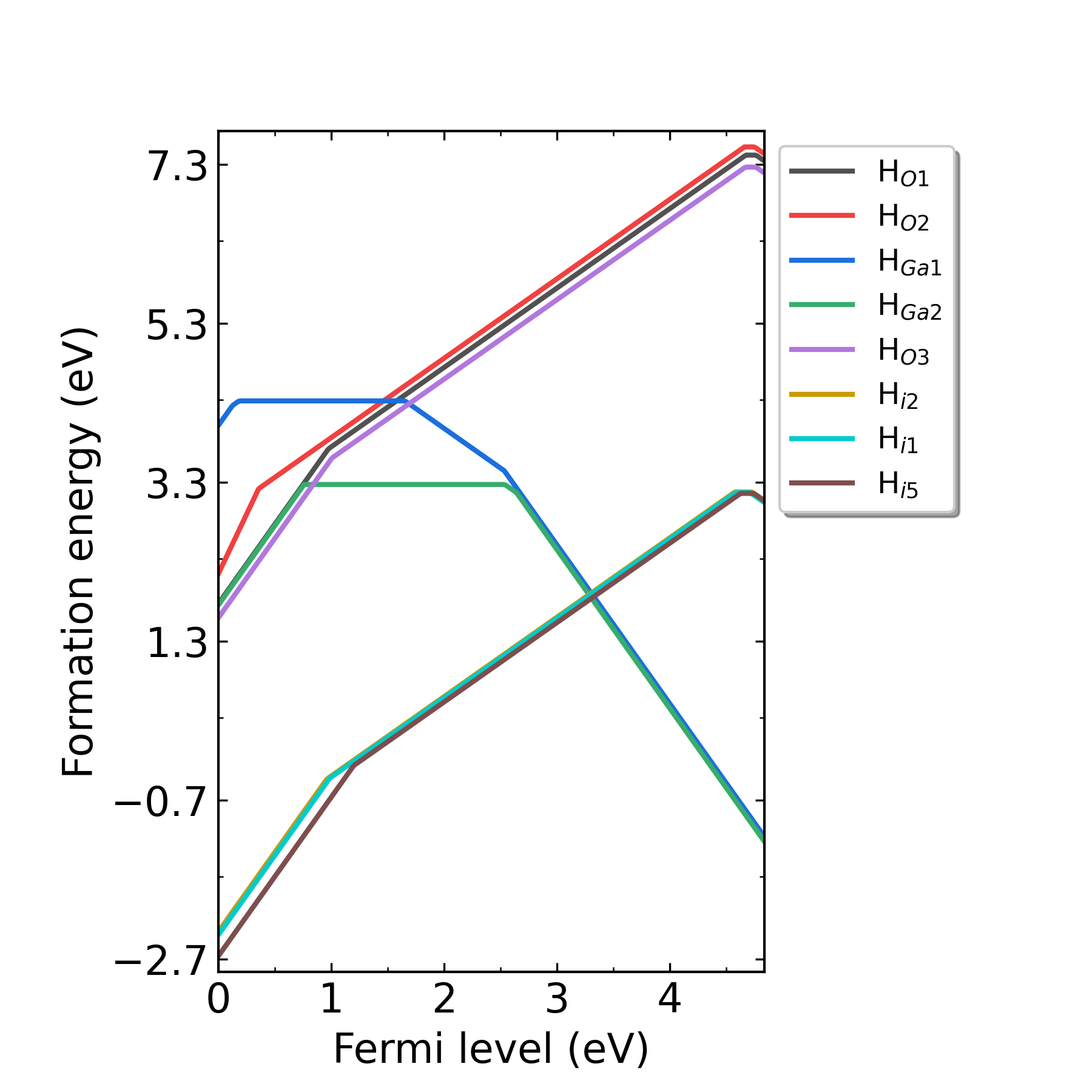
掺杂H的Ga2O3在p2处(O-rich)的缺陷形成能随费米能级的变化。
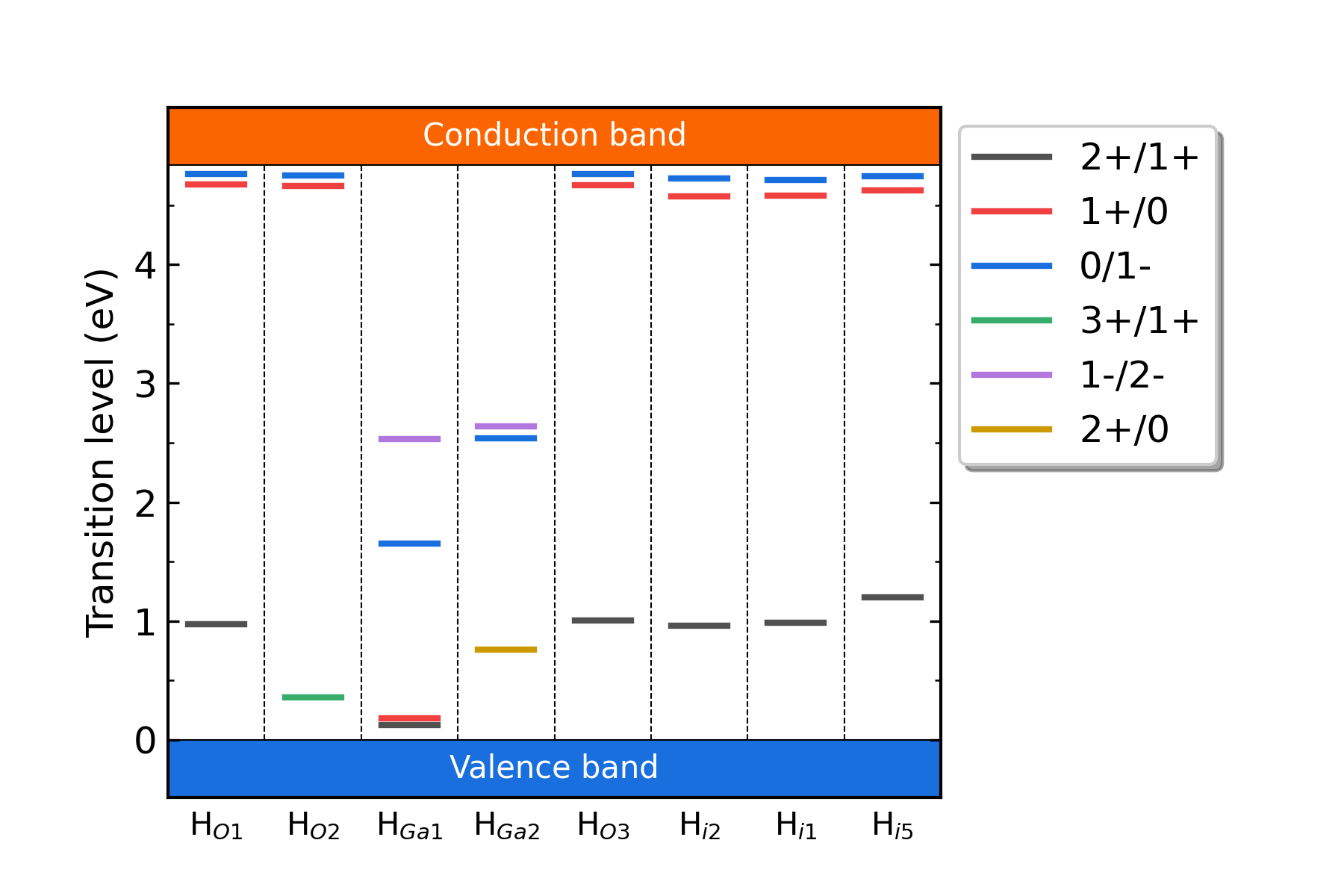
掺杂H的Ga2O3各缺陷的转变能级。
5.3.4. 缺陷浓度和费米能级计算DDC¶
5.3.4.1. 运行DDC模块¶
5.3.4.2. DDC模块运行流程¶
缺陷数据汇总:
DefectParams.txt 文件中。4ddc.out :############ Collecting information from DEC ############
Read defect types from DEC calculation successfully.
Defects considered in DDC calculation: ['H_O1', 'H_O2', 'H_Ga1', 'H_Ga2', 'H_O3', 'H_i-2', 'H_i-1', 'H_i-5']
Chemical potentials change from p1 to p2.
Calculate gq for defect in each charge state.
Calculate Nsites for H_O1: 5.727808e+22 cm^-3.
Calculate Nsites for H_O2: 5.727808e+22 cm^-3.
Calculate Nsites for H_Ga1: 3.818539e+22 cm^-3.
Calculate Nsites for H_Ga2: 3.818539e+22 cm^-3.
Calculate Nsites for H_O3: 5.727808e+22 cm^-3.
Calculate Nsites for H_i-2: 3.182116e+22 cm^-3.
Calculate Nsites for H_i-1: 3.182116e+22 cm^-3.
Calculate Nsites for H_i-5: 3.182116e+22 cm^-3.
############ Collecting information from DEC ############
DefectParams.txt 文件:1000 300
0.230000 4.210000
4.836382
H_O1 5.727808e+22 2 4.818 1 2.627 2 1.739 1 x x 4.769 1 x x x x x x 1.840000 7.420000
H_O2 5.727808e+22 2 4.812 1 2.461 2 1.811 1 1.36 2 4.768 1 x x x x x x 1.942000 7.522000
H_Ga1 3.818539e+22 1 0.466 2 0.373 1 0.332 2 0.336 1 1.41 2 1.647 1 2.779 2 3.312 1 8.045000 4.325000
H_Ga2 3.818539e+22 1 0.857 2 0.756 1 0.571 2 0.374 1 2.307 2 2.163 1 3.112 2 3.554 1 6.993000 3.272000
H_O3 5.727808e+22 2 4.815 1 2.622 2 1.736 1 1.287 2 4.771 1 x x x x x x 1.688000 7.268000
H_i-2 3.182116e+22 2 4.738 1 2.571 2 1.708 1 1.266 2 4.733 1 x x x x x x 1.318000 3.178000
H_i-1 3.182116e+22 2 4.747 1 2.577 2 1.712 1 1.233 2 4.719 1 x x x x x x 1.311000 3.171000
H_i-5 3.182116e+22 2 4.779 1 2.581 2 1.592 1 1.23 2 4.741 1 x x x x x x 1.301000 3.160000
生长温度下自洽计算:
DDC模块在T=1000 K的时候计算其缺陷浓度和载流子浓度,并根据电中性条件自洽求解费米能级。
工作温度下自洽计算:
DDC模块在T=300 K的时候重新分布每个缺陷各价态的浓度,并根据电中性条件再次自洽求解费米能级。
输出缺陷浓度:
DDC模块在doping-Ga2O3/ddc目录下,,输出三个文件:费米能级文件 Fermi.dat ,载流子浓度文件 Carrier.dat ,缺陷浓度文件 Defect_charge.dat 。可使用Origin画图。
此外,DDC模块会自动根据三个文件画图,产生density.png文件。如下图所示:
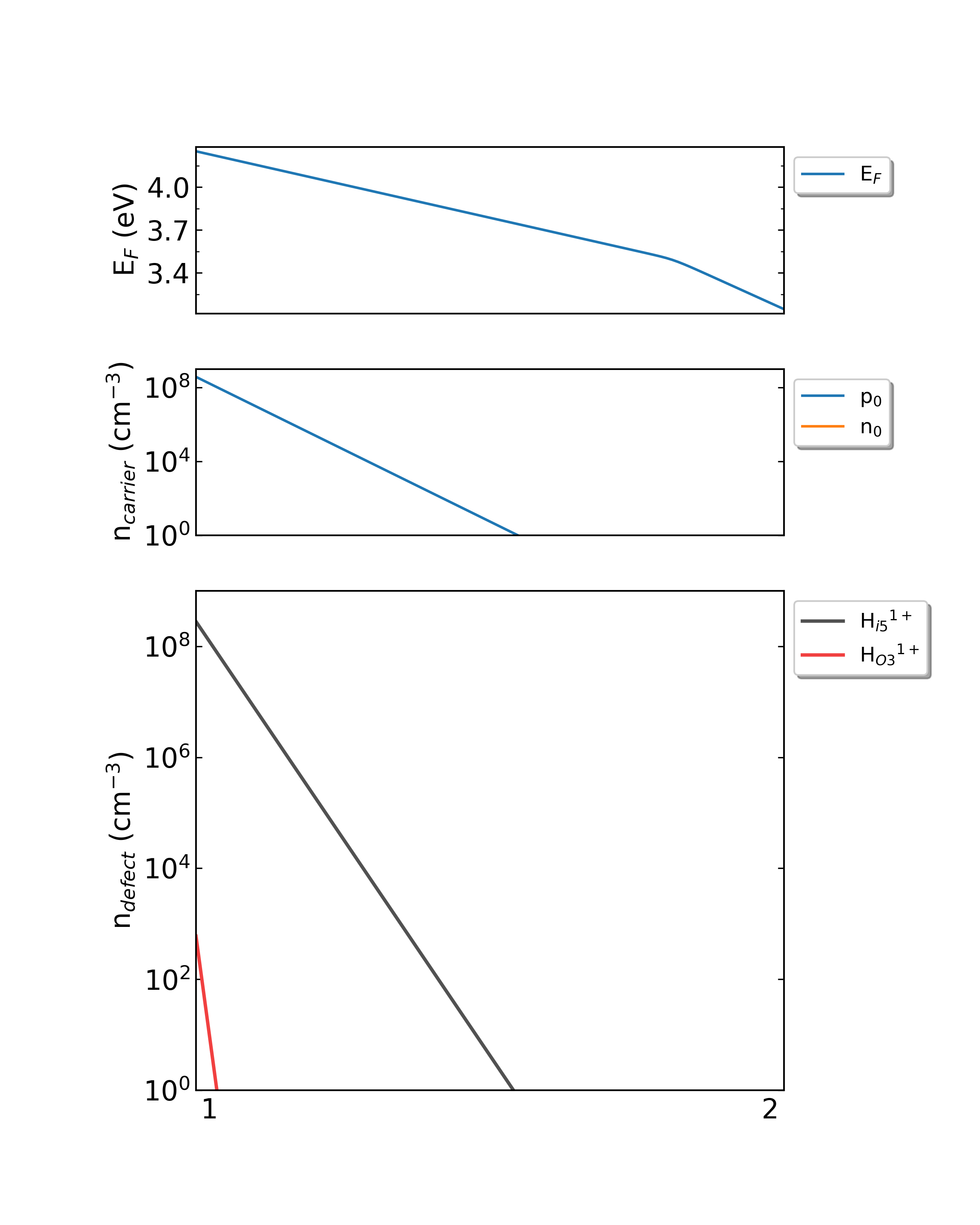
生长温度为300K时,掺杂H的Ga2O3从p1(Ga-rich)到p2(O-rich)的费米能级、载流子浓度、缺陷浓度。
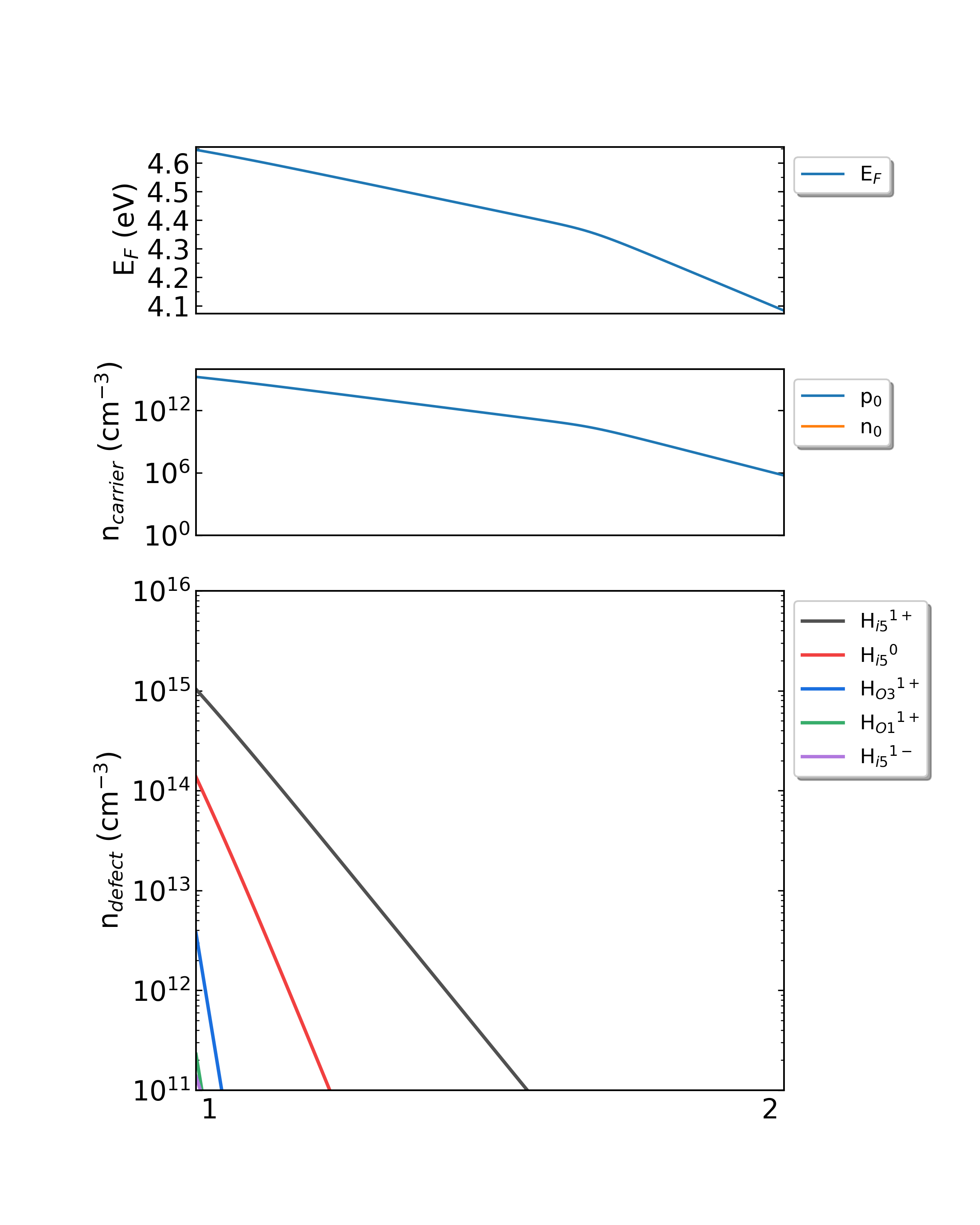
生长温度为650K时,掺杂H的Ga2O3从p1(Ga-rich)到p2(O-rich)的费米能级、载流子浓度、缺陷浓度。
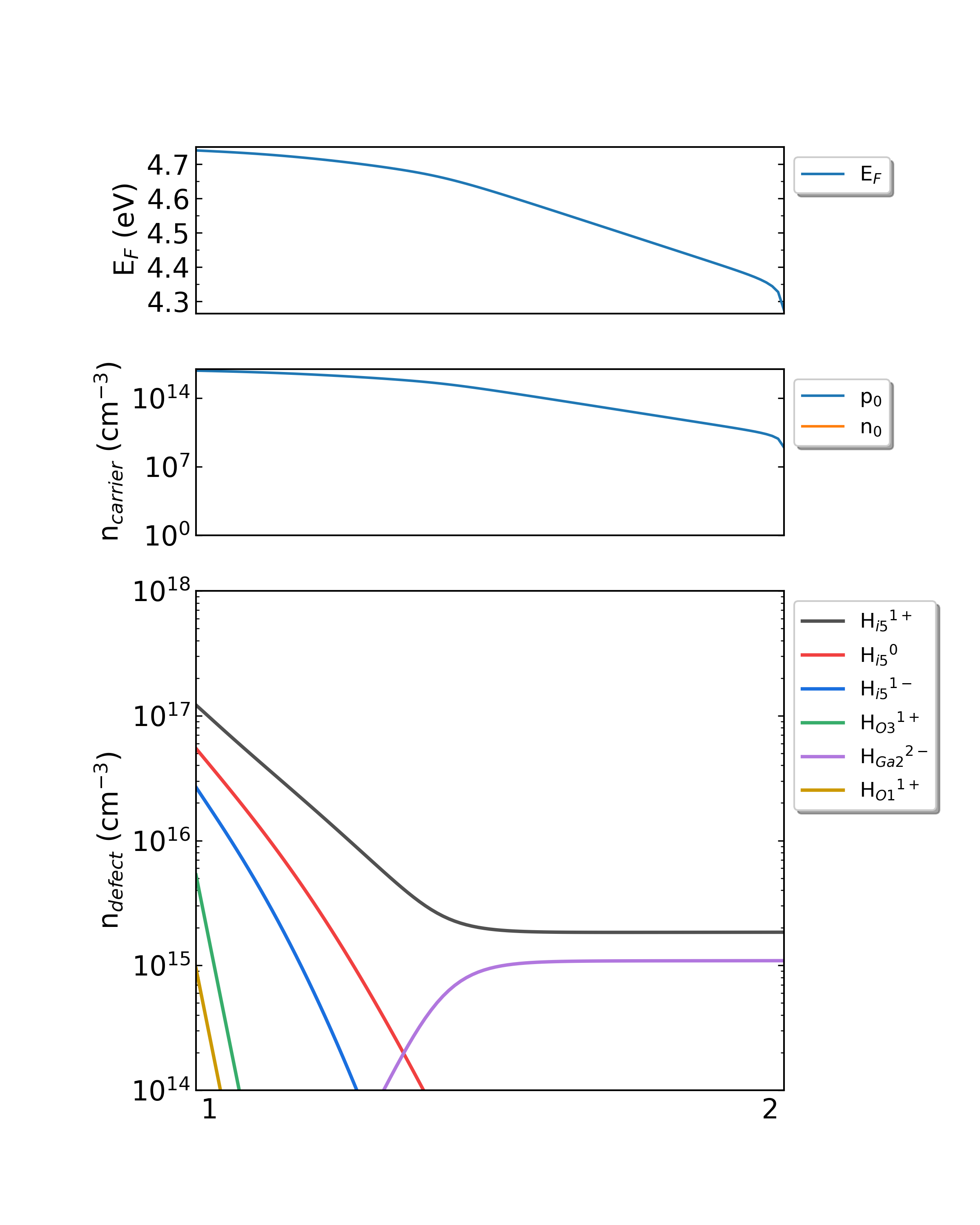
生长温度为1000K时,掺杂H的Ga2O3从p1(Ga-rich)到p2(O-rich)的费米能级、载流子浓度、缺陷浓度。
5.4. ZnGeP2的本征缺陷计算¶
5.4.1. 准备计算PREPARE¶
5.4.1.1. 准备POSCAR与dasp.in¶
POSCAR,使用VASP优化其晶格常数,或修改其晶格常数从而匹配实验值(此步骤由用户自行完成)。显示如下:Zn4 Ge4 P8
1.0
5.468 0.0000000000 0.0000000000
0.0000000000 5.468 0.0000000000
0.0000000000 0.0000000000 10.745
Zn Ge P
4 4 8
Direct
0.000000000 0.500000000 0.250000000
0.000000000 0.000000000 0.000000000
0.500000000 0.000000000 0.750000000
0.500000000 0.500000000 0.500000000
0.500000000 0.000000000 0.250000000
0.500000000 0.500000000 0.000000000
0.000000000 0.500000000 0.750000000
0.000000000 0.000000000 0.500000000
0.250000000 0.754127026 0.375000000
0.745872974 0.750000000 0.125000000
0.254126996 0.250000000 0.125000000
0.750000000 0.245873004 0.375000000
0.750000000 0.254126996 0.875000000
0.245873004 0.250000000 0.625000000
0.754127026 0.750000000 0.625000000
0.250000000 0.745872974 0.875000000
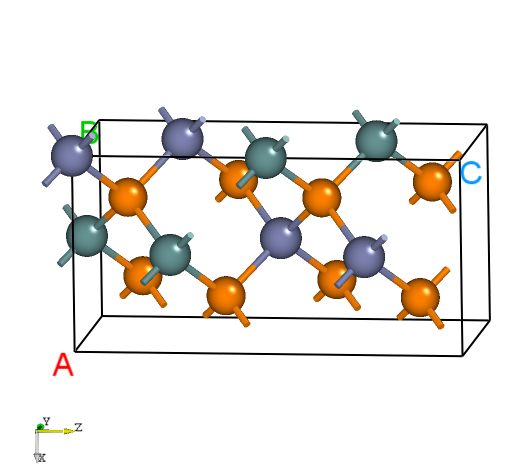
ZnGeP2的晶体结构。
############## Job Scheduling ##############
cluster = SLURM # (job scheduling system)
node_number = 4 # (number of node)
core_per_node = 32 # (core per node)
queue = normal # (name of queue/partition)
max_time = 24:00:00 # (maximum time for a single DFT calculation)
vasp_path_dec = /opt/vasp.5.4.4/bin/vasp_gam # (path of VASP)
vasp_path_tsc = /opt/vasp.5.4.4/bin/vasp_std
vasp_path_cdc=/opt/vasp-optics/bin/vasp_gam
job_name = submit_job # (name of script)
potcar_path = /opt/POT/potpaw_PBE # (path of pseudopotentials)
max_job = 5
############## TSC Module ##############
database_api = ******************* # (str-list type)
############## DEC Module ##############
level = 2 # (level=1: PBE+PBE; level=2: PBE+HSE; level=3: HSE+HSE)
min_atom = 180
max_atom = 200
intrinsic = T # (default: T)
correction = FNV # (default: none)
epsilon = 12.3
Eg_real = 2.06 # (experimental band gap)
############## DDC Module ##############
ddc_temperature = 1300 300
ddc_mass = 0.36 0.54
ddc_path = 1 2
############## CDC Module ###############
cdc_defect = Ge_Zn1
cdc_job = pl / radiative_rate
cdc_charge = 0 1
cdc_band = 864 865
cdc_temperature = 300
spin_channel = 2
refractive_index = 2.38
cluster = SLURM
# 表示使用集群的队列系统为SLURM
node_number = 4
# 对于每个单独的计算,使用4个节点
core_per_node = 32
# 对于每个节点,使用32个核。因此对于每个单独的计算,总共使用4*32=128核
queue = normal
# 使用名为"normal"的队列进行计算。因此,在设置dasp.in参数之前,需要确认超算/集群上的队列名、节点、核数
max_time = 24:00:00 # (maximum time for a single DFT calculation)
# 每个单独的计算所允许的最大时间为24小时,可任意设置。
vasp_path_dec = /opt/vasp.5.4.4/bin/vasp_gam # (path of VASP)
vasp_path_tsc = /opt/vasp.5.4.4/bin/vasp_std
vasp_path_cdc = /opt/vasp-optics/bin/vasp_gam
# 对于TSC的计算,采用std版的VASP。对于DEC的单k点计算,采用gam版的VASP。CDC计算中会调用指定gam版的VASP计算载流子跃迁矩阵元。
job_name = submit_job # (name of script)
# 提交任务的脚本,命名为"submit_job",可任意设置。
potcar_path = /opt/POT/potpaw_PBE # (path of pseudopotentials)
# POTCAR的路径
max_job = 5
# 允许同时在跑的任务最大数
database_api = ******************* # (str-list type)
# 用于访问Materials Project数据库
level = 2 # (level=1: PBE+PBE; level=2: PBE+HSE; level=3: HSE+HSE)
# 对于超胞,使用PBE优化其原子位置,使用HSE计算其总能
min_atom = 180
max_atom = 200
# 我们希望生成的超胞大小在180-200个原子之间,且尽量使a=b=c,a⊥b⊥c
intrinsic = T # (default: T)
# 产生本征缺陷,V_Zn V_Ge V_P Zn_Ge Zn_P Ge_Zn Ge_P P_Zn P_Ge Zn_i Ge_i P_i
correction = FNV # (default: none)
# 带电缺陷的修正方案采用FNV修正
epsilon = 12.3
# ZnGeP2的介电常数为12.3
Eg_real = 2.06 # (experimental band gap)
# ZnGeP2的实验带隙值约为2.06 eV,DASP将根据此数据调整AEXX参数,从而使得无缺陷超胞的带隙值等于2.06 eV
ddc_temperature = 1300 300
# 设置生长温度为1300 K,工作温度为300 K
ddc_mass = 0.36 0.54
# 设置电子有效质量为0.36,空穴有效质量为0.54
ddc_path = 1 2
# 设置DDC计算的化学势路径,1 2 对应dasp.in中第一个化学势(p1)到第二个化学势(p2)
cdc_defect = Ge_Zn1
# 计算Ge_Zn1缺陷的相关性质
cdc_job = pl / radiative_rate
# 计算PL谱/辐射俘获系数
cdc_charge = 0 1
# 缺陷态从中性态转为电荷量为+q的电离态,为空穴跃迁
cdc_band = 864 865
# 空穴载流子跃迁,从价带顶跃迁至缺陷态上,即第864条能带跃迁至第865条能带
cdc_temperature = 300
# CDC模块计算300K下的缺陷性质
spin_channel = 2
# 载流子自旋向下
refractive_index = 2.38
# 材料的折射率为2.38
5.4.1.2. 使用DASP产生必要输入文件¶
dasp.in 文件,执行 dasp 1 ,即可启动PREPARE模块,此后无需额外操作。DASP会输出 1prepare.out 文件记录程序的运行日志。5.4.1.3. PREPARE模块运行流程¶
产生超胞:
POSCAR 文件。以下为ZnGeP2原胞扩成的超胞 POSCAR_nearlycube :Cubic_cell
1.0
16.4040000000 0.0000000000 0.0000000000
0.0000000000 15.3313770093 0.0000000000
0.0000000000 0.2701043094 15.3289975100
Zn Ge P
48 48 96
Direct
0.0000000000 1.0000000000 0.2500000000
0.0000000000 0.0000000000 0.0000000000
0.1666666666 0.6250000000 0.3750000000
0.1666666666 0.8750000000 0.3750000000
...
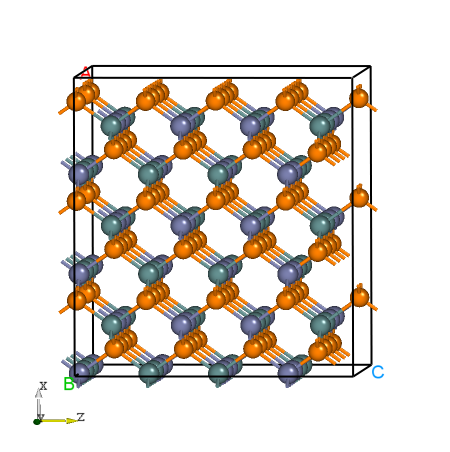
ZnGeP2的超胞结构。
马德隆常数计算:
1prepare.out 的输出如下(其中********表示该任务的计算id):############ Prepare Files module start ############
Read the structure file POSCAR you provided
Get the refined cell POSCAR_refined from POSCAR
Generate the nearlycube cell POSCAR_nearlycube from POSCAR
Generate job script through dasp.in parameters
Generate single-point KPOINTS
Generate pseudopotential file POTCAR through potcar_dir you set
Generate commonly used vasp input file INCAR
Start the madelung constant calculation
Generate the madelung calculation directory
Generate madelung calculation POSCAR
Generate madelung calculation POTCAR
Generate madelung calculation INCAR
Generate madelung calculation KPOINTS
Generate madelung calculation job script
Job ******** submitted: /home/test/ZnGeP2/dec/madelung/static
Succeed job ********: /home/test/ZnGeP2/dec/madelung/static
The madelung constant calculation completed
The madelung constant = 2.833
HSE交换参数计算:
cd ./dec/AEXX
ls
0.25 0.26795555051593156 0.3 AEXX.list
INCAR。同时从 1prepare.out 可以看到如下日志(其中********表示该任务的计算id):Start the HSE parameter AEXX calculation
Job ******** submitted: /home/test/ZnGeP2/dec/AEXX/0.25/static
Job ******** submitted: /home/test/ZnGeP2/dec/AEXX/0.3/static
Succeed job ********: /home/test/ZnGeP2/dec/AEXX/0.25/static
Succeed job ********: /home/test/ZnGeP2/dec/AEXX/0.3/static
Job ******** submitted: /home/test/ZnGeP2/dec/AEXX/0.26795555051593156/static
Succeed job ********: /home/test/ZnGeP2/dec/AEXX/0.26795555051593156/static
The HSE parameter AEXX calculation completed
The HSE parameter AEXX = 0.27
level = 2: Generate PBE relax vasp input file INCAR-relax
level = 2: Generate HSE static vasp input file INCAR-static
host超胞原子位置的优化:
1prepare.out 可以DASP运行结束的标志,并告诉我们下一步需要做TSC模块的计算(其中********表示该任务的计算id)。Start the POSCAR_nearlycube relax calculation
Generate the POSCAR_nearlycube relax directory
Job ******** submitted: /home/test/ZnGeP2/dec/relax
Succeed job ********: /home/test/ZnGeP2/dec/relax
The POSCAR_nearlycube relax calculation completed
Get the final structure POSCAR_final
############ Prepare Files module end ############
DASP-PREPARE finished, please run DASP-TSC next
5.4.2. 热力学稳定性和元素化学势计算TSC¶
5.4.2.1. 运行TSC模块¶
dasp 1 执行PREPARE模块时,会生成ZnGeP2/dec目录,并在该目录中产生 1prepare.out 文件。等待程序执行完毕, 1prepare.out 有相应的完成标志。进入ZnGeP2/dec目录。确认INCAR-relax,INCAR-static文件中的参数是可行的。(用户可修改INCAR,DASP将根据此目录中的INCAR做后续的计算)dasp 2 执行TSC模块。同样地,TSC模块会在ZnGeP2目录中生成名为tsc的目录,里面记录了TSC程序的计算输出,包括各计算目录以及运行日志文件 1prepare.out 。等待程序完成期间无需额外操作。5.4.2.2. TSC模块运行流程¶
host结构的总能计算(与MP参数保持一致):
TSC模块将使用与 Materials Project 数据库提供的输入参数(INCAR,KPOINTS,POTCAR)来对用户给定的原胞做结构优化和静态计算,该计算得到的总能与MP数据库的总能是可比的。此步骤是为了得到影响ZnGeP2稳定性的 关键杂相 。通过目录可以看到:
cd tsc
cd ZnGeP2/
ls
relaxation1 relaxation2 static
从ZnGeP2/tsc/2tsc.out中也可以看到程序的运行日志,即产生输入文件、relaxation1、relaxation2、static、数据提取等步骤。
关键杂相判断:
TSC模块将搜寻MP数据库上所有与ZnGeP2相竞争的杂项,通过DFT计算的ZnGeP2的总能与MP数据库中杂相的总能,判断出ZnGeP2是 稳定的 。
随后,程序将计算获取影响ZnGeP2稳定性最关键的杂相,本例中包括Ge,P,Zn3P2,ZnP2和Zn。在 2tsc.out 中可看到相关的信息:
...
analysing the thermodynamic stability of ZnGeP2.
key phases of ZnGeP2 are: Ge P Zn3P2 ZnP2 Zn .
file key_phases_info_recalc.yaml generated.
analysing of ZnGeP2 is done.
...
host与杂相结构的总能计算(PREPARE模块确定的参数):
在确定关键杂相后,TSC模块将使用PREPARE模块确定的参数(AEXX)计算ZnGeP2,Ge,P,Zn3P2,ZnP2和Zn的总能。 2tsc.out 如下:
...
Job ******** submitted: /home/test/ZnGeP2/tsc/ZnGeP2/static_recalc
Job ******** submitted: /home/test/ZnGeP2/tsc/Ge/static_recalc
Job ******** submitted: /home/test/ZnGeP2/tsc/P/static_recalc
Job ******** submitted: /home/test/ZnGeP2/tsc/Zn3P2/static_recalc
Job ******** submitted: /home/test/ZnGeP2/tsc/ZnP2/static_recalc
Job ******** submitted: /home/test/ZnGeP2/tsc/Zn/static_recalc
Succeed job ********: /home/test/ZnGeP2/tsc/ZnGeP2/static_recalc
Succeed job ********: /home/test/ZnGeP2/tsc/Ge/static_recalc
Succeed job ********: /home/test/ZnGeP2/tsc/P/static_recalc
Succeed job ********: /home/test/ZnGeP2/tsc/Zn3P2/static_recalc
Succeed job ********: /home/test/ZnGeP2/tsc/Zn/static_recalc
Succeed job ********: /home/test/ZnGeP2/tsc/ZnP2/static_recalc
...
化学势的计算:
根据DFT计算的总能,计算ZnGeP2的形成能和化学势稳定区间,TSC模块给出4个化学势的端点值,写入 dasp.in :
# 顺序与POSCAR中元素顺序一致,即第一列是Zn,第二列是Ge,第三列是P。
E_pure = -2.0283 -5.9739 -7.3365
p1 = -0.1456 0.0 -0.4672
p2 = -1.08 0.0 0.0
p3 = -0.9207 -0.1593 0.0
p4 = -0.2252 -0.1593 -0.3478
在 2tsc.out 可以看到程序执行完毕的输出:
dir '2d-figures','3d-figures','ori_data_MP' ready. try to read file: 'calc_list.yaml'.
analysing the thermodynamic stability of ZnGeP2.
key phases of ZnGeP2 are: Ge P Zn3P2 ZnP2 Zn .
analysing of ZnGeP2 is done.
DASP-TSC finished
对于三元与四元的化合物,TSC模块将输出稳定区域图像,及稳定区域各端点处的化学势。通过目录可以看到:
cd tsc
cd 2d-figures/
ls
fig-ZnGeP2.png fig-ZnGeP2_recalc.png stable_2d.out stable_recalc_2d.out
目录ZnGeP2/tsc/2d-figures/中的四个文件分别是两次计算与分析过程中绘制的稳定区域图像以及图像中各端点处的化学势。
查看文件 stable_2d.out 与 fig-ZnGeP2.png 。图 fig-ZnGeP2.png 的横纵坐标分别是图中所标识元素的化学势,阴影区域
则是目标化合物的稳定区域,其边界的每一条线
是相应所标识材料恰好处于形成与未形成的临界情况下的化学势曲线,这是第一次计算与分析过程输出的图像。
Ge Zn P
------- ------- -------
-0.0999 -0.7457 0
0 -0.8457 0
0 -0.0484 -0.3986
-0.0999 -0.0983 -0.3237
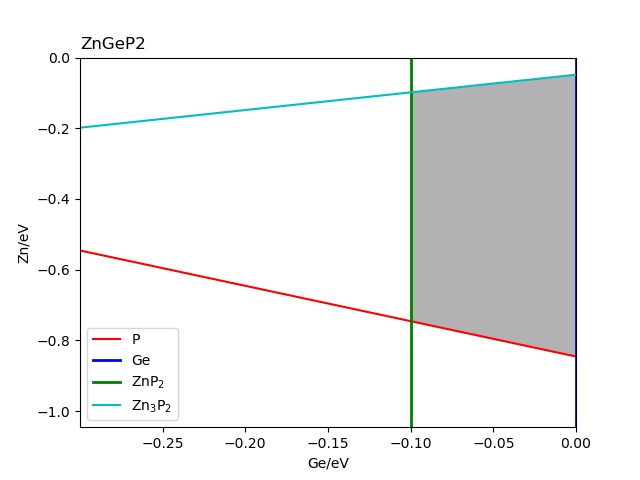
ZnGeP2的稳定区域图(来自MP数据库)。
查看文件 stable_recalc_2d.out 与 fig-ZnGeP2_recalc.png ,这是第二次计算与分析过程输出的数据与图像。
Ge Zn P
------- ------- -------
-0.1593 -0.9207 0
0 -1.08 0
0 -0.1456 -0.4672
-0.1593 -0.2252 -0.3478
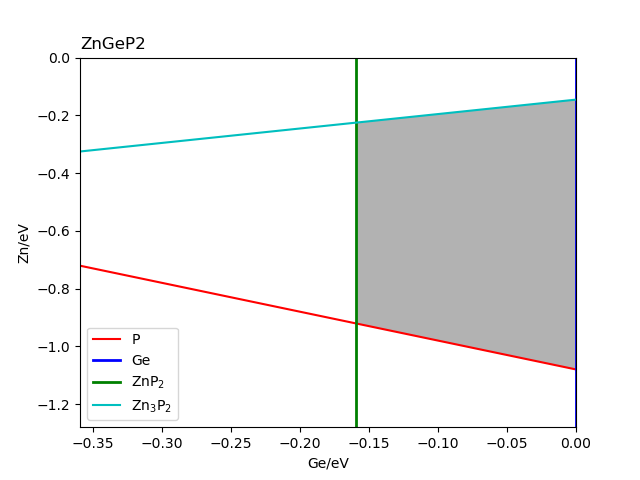
ZnGeP2的稳定区域图(来自DFT计算)。
5.4.3. 缺陷形成能和转变能级计算DEC¶
5.4.3.1. 运行DEC模块¶
在上一步使用命令 dasp 2 执行TSC模块时,会生成ZnGeP2/tsc目录,并在该目录中产生 2tsc.out 文件。等待程序执行完毕, 2tsc.out 有相应的完成标志。打开ZnGeP2/dasp.in,确认化学势已被程序自动输入。
确认TSC模块完成后,回到ZnGeP2目录,使用命令 dasp 3 执行DEC模块。DEC模块会在第一步已经生成的dec目录中继续输出相关文件,包括缺陷结构,缺陷计算目录,以及运行日志文件 3dec.out 。等待程序完成期间无需额外操作。
5.4.3.2. DEC模块运行流程¶
产生缺陷结构:
为计算得到材料的缺陷性质,程序将根据用户在 dasp.in 中设置需要计算的缺陷类型生成相应的计算目录以及电中性的缺陷构型。
cd dec
cd Intrinsic_Defect
ls
Ge_i Ge_Zn1 Intrinsic_Defect.list P_i V_Ge1 V_Zn1 Zn_i
Ge_P1 host P_Ge1 P_Zn1 V_P1 Zn_Ge1 Zn_P1
从文件 3dec.out 中可以看到如下日志:
############ Neutral Defect module start ############
Make intrinsic defect directory Intrinsic_Defect
Generate host directory in Intrinsic_Defect
Start generating neutral vacancy defect
Generate neutral defect at: V_Zn1/initial_structure/q0
Generate neutral defect at: V_Ge1/initial_structure/q0
Generate neutral defect at: V_P1/initial_structure/q0
Neutral vacancy defect generation completed
Start generating neutral intrinsic antisite defect
Generate neutral defect at: Ge_Zn1/initial_structure/q0
Generate neutral defect at: P_Zn1/initial_structure/q0
Generate neutral defect at: Zn_Ge1/initial_structure/q0
Generate neutral defect at: P_Ge1/initial_structure/q0
Generate neutral defect at: Zn_P1/initial_structure/q0
Generate neutral defect at: Ge_P1/initial_structure/q0
Neutral intrinsic antisite defect generation completed
Start generating neutral intrinsic interstitial defect
Generate neutral defect at: Zn_i/random1/initial_structure/q0
Generate neutral defect at: Zn_i/random2/initial_structure/q0
...
Generate neutral defect at: Ge_i/random1/initial_structure/q0
Generate neutral defect at: Ge_i/random2/initial_structure/q0
...
Generate neutral defect at: P_i/random1/initial_structure/q0
Generate neutral defect at: P_i/random2/initial_structure/q0
...
Neutral intrinsic interstitial defect generation completed
############ Neutral Defect module end ############
产生的各类缺陷结构中,部分反位缺陷与可能的间隙位如下图所示:
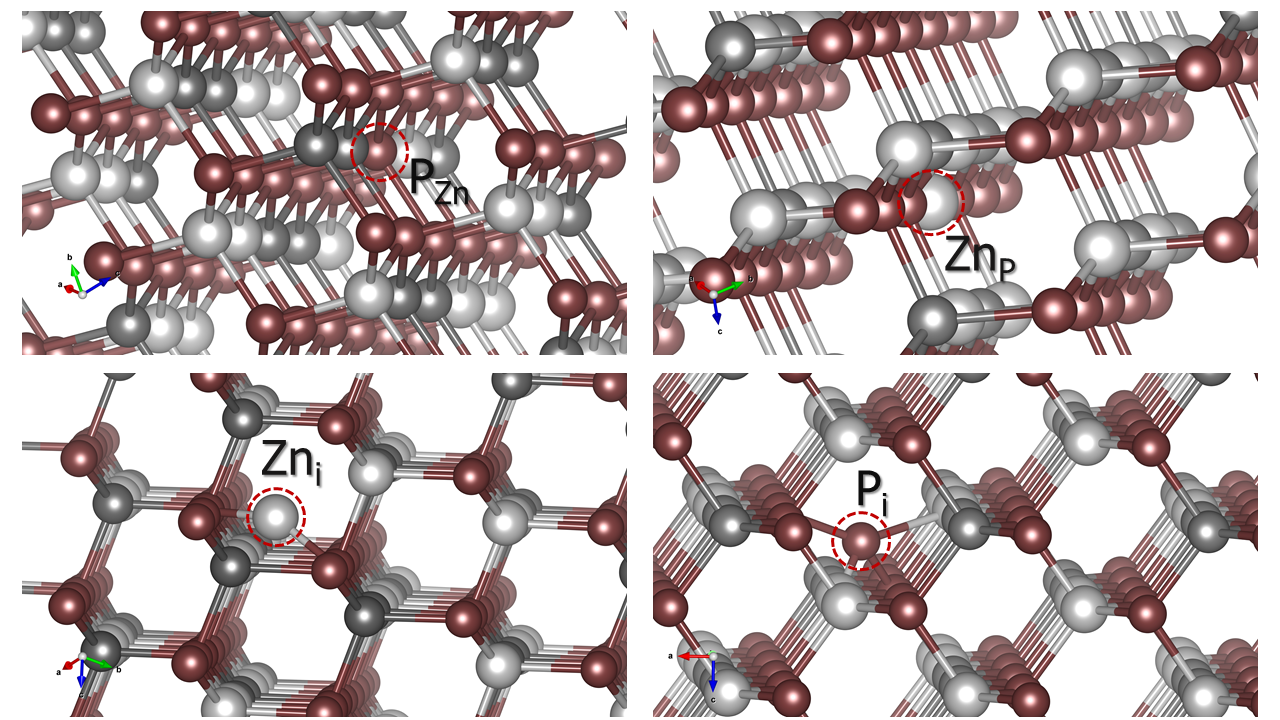
ZnGeP2超胞部分缺陷结构示意图。
提交各缺陷q=0计算任务:
在建立好各类型的电中性缺陷计算目录与构型后,程序将自动补充第一性原理计算所需文件并依次提交计算任务,并使得同时计算的任务数不超过 dasp.in 中参数 max_job 的值。从文件 3dec.out 中可以看到如下日志:
############ AutoRun - Neutral Defect module start ############
Job ******** submitted: /data/home/test/Zn_Ge_P/DASP-test-0128/dec/Intrinsic_Defect/P_Zn1/initial_structure/q0
Job ******** submitted: /data/home/test/Zn_Ge_P/DASP-test-0128/dec/Intrinsic_Defect/Zn_P1/initial_structure/q0
Job ******** submitted: /data/home/test/Zn_Ge_P/DASP-test-0128/dec/Intrinsic_Defect/Ge_P1/initial_structure/q0
Job ******** submitted: /data/home/test/Zn_Ge_P/DASP-test-0128/dec/Intrinsic_Defect/P_Ge1/initial_structure/q0
Job ******** submitted: /data/home/test/Zn_Ge_P/DASP-test-0128/dec/Intrinsic_Defect/P_i/random3/initial_structure/q0
Succeed job ********: /data/home/test/Zn_Ge_P/DASP-test-0128/dec/Intrinsic_Defect/Zn_P1/initial_structure/q0
Job ******** submitted: /data/home/test/Zn_Ge_P/DASP-test-0128/dec/Intrinsic_Defect/P_i/random1/initial_structure/q0
Succeed job ********: /data/home/test/Zn_Ge_P/DASP-test-0128/dec/Intrinsic_Defect/Ge_P1/initial_structure/q0
...
############ AutoRun - Neutral Defect module end ############
产生带电缺陷的计算目录:
在中性缺陷计算完成后,程序将判断各缺陷可能的电离态数量,并进一步生成各电离态的计算目录与缺陷构型。
############ Ionized Defect module start ############
Start generating ionized defects
Ionized defect path: /data/home/test/Zn_Ge_P/DASP-test-0128/dec/Intrinsic_Defect/P_Zn1/initial_structure/q-4
Ionized defect path: /data/home/test/Zn_Ge_P/DASP-test-0128/dec/Intrinsic_Defect/P_Zn1/initial_structure/q-3
...
Ionized defects generation completed
############ Ionized Defect module end ############
提交各缺陷q≠0的计算任务:
在建立好各类型的电离态缺陷计算目录与构型后,程序将自动补充第一性原理计算所需文件并依次提交计算任务,并使得同时计算的任务数不超过 dasp.in 中参数 max_job 的值。从文件 3dec.out 中可以看到如下日志:
############ AutoRun - Ionized Defect module start ############
Job ******** submitted: /data/home/test/Zn_Ge_P/DASP-test-0128/dec/Intrinsic_Defect/P_Zn1/initial_structure/q-1
Job ******** submitted: /data/home/test/Zn_Ge_P/DASP-test-0128/dec/Intrinsic_Defect/P_Zn1/initial_structure/q-4
Job ******** submitted: /data/home/test/Zn_Ge_P/DASP-test-0128/dec/Intrinsic_Defect/P_Zn1/initial_structure/q-2
Job ******** submitted: /data/home/test/Zn_Ge_P/DASP-test-0128/dec/Intrinsic_Defect/P_Zn1/initial_structure/q+3
Job ******** submitted: /data/home/test/Zn_Ge_P/DASP-test-0128/dec/Intrinsic_Defect/P_Zn1/initial_structure/q-3
Succeed job ********: /data/home/test/Zn_Ge_P/DASP-test-0128/dec/Intrinsic_Defect/P_Zn1/initial_structure/q-1
Job ******** submitted: /data/home/test/Zn_Ge_P/DASP-test-0128/dec/Intrinsic_Defect/P_Zn1/initial_structure/q+1
...
############ AutoRun - Ionized Defect module end ############
计算带电缺陷的修正:
在各类型的电离态缺陷计算完成后,程序将计算各类型缺陷的形成能、各电离态的转变能级,在日志文件 3dec.out 中记录有各类缺陷在不同化学势情况下的形成能、能带对齐和镜像电荷修正项(LZ/FNV),以及不同电离态的转变能级。 dasp.in 文件中提供了四种各元素的化学势取值情况,因此有p1,p2,p3,p4四种形成能值。从文件 3dec.out 中可以看到如下日志:
############ Formation Energy module start ############
Start the formation energy calculation
The formation energy (neutral) of P_Zn1 at p1 is 3.993075
The formation energy (neutral) of P_Zn1 at p2 is 2.591475
The formation energy (neutral) of P_Zn1 at p3 is 2.750775
The formation energy (neutral) of P_Zn1 at p4 is 3.794075
The FNV correction (q = -1) E_correct = 0.0859854 eV
The transition level (-/0) above VBM: 1.3758
The FNV correction (q = -4) E_correct = 1.43843 eV
The transition level (4-/0) above VBM: 2.0373
...
############ Formation Energy module end ############
输出形成能图像:
- 在各类型缺陷形成能与转变能级的计算完成后,程序将自动生成在不同化学势情况下的缺陷形成能图像及数据,存于目录/dec/Formation_Energy_Intrinsic_Defect/中。
dasp.in文件中提供了四种各元素的化学势取值情况,因此有四份图像与数据。
cd dec
cd Formation_Energy_Intrinsic_Defect/
ls
p1.dat p1.png p2.dat p2.png p3.dat p3.png p4.dat p4.png
用户可根据.dat文件中的数据自行绘制图像,或参考程序自动绘制的.png图像文件。从文件 3dec.out 中可以看到如下日志:
############ Plot Diagram module start ############
Start plotting the diagrams
Generate formation energy diagrams at p1: /data/home/test/Zn_Ge_P/DASP-test-0128/dec/Formation_Energy_Intrinsic_Defect/p1.dat
Generate formation energy diagrams at p2: /data/home/test/Zn_Ge_P/DASP-test-0128/dec/Formation_Energy_Intrinsic_Defect/p2.dat
Generate formation energy diagrams at p3: /data/home/test/Zn_Ge_P/DASP-test-0128/dec/Formation_Energy_Intrinsic_Defect/p3.dat
Generate formation energy diagrams at p4: /data/home/test/Zn_Ge_P/DASP-test-0128/dec/Formation_Energy_Intrinsic_Defect/p4.dat
Generate transition level diagram: /data/home/test/Zn_Ge_P/DASP-test-0128/dec/Transition_Level_Intrinsic_Defect/tl.dat
All diagrams completed
############ Plot Diagram module end ############
程序自动绘制的四幅图像分别如下所示
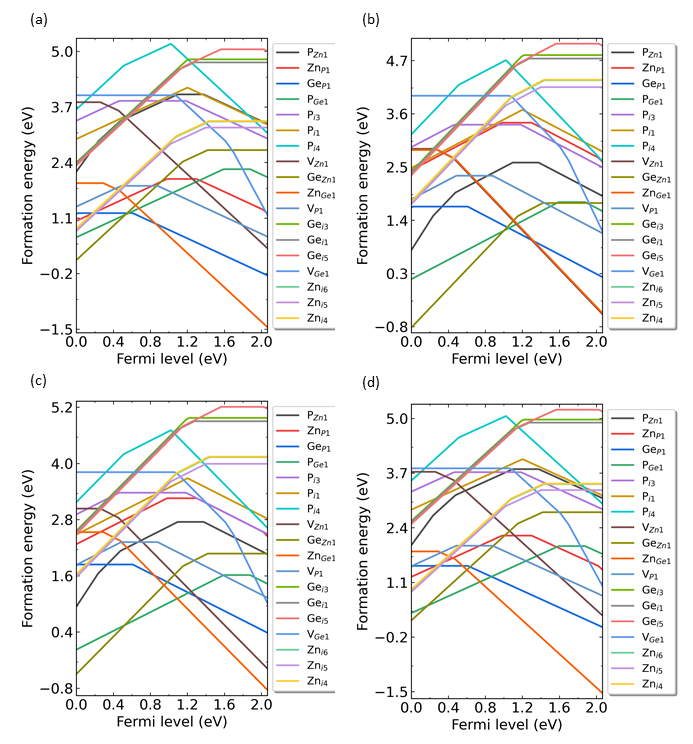
ZnGeP2各类缺陷在化学势点(a) p1, (b) p2, (c) p3, (d) p4 处的形成能随费米能级的变化。
5.4.4. 缺陷浓度和费米能级计算DDC¶
5.4.4.1. 运行DDC模块¶
dasp 4 执行DDC模块。等待期间无需额外操作。5.4.4.2. DDC模块运行流程¶
缺陷数据汇总:
DefectParams.txt 文件中。4ddc.out :############ Collecting information from DEC ############
Read defect types from DEC calculation successfully.
Defects considered in DDC calculation: ['P_Zn1', 'Zn_P1', 'Ge_P1', 'P_Ge1', 'P_i-3', 'P_i-1', 'P_i-4', 'V_Zn1', 'Ge_Zn1', 'Zn_Ge1', 'V_P1', 'Ge_i-3', 'Ge_i-1', 'Ge_i-5', 'V_Ge1', 'Zn_i-6', 'Zn_i-5', 'Zn_i-4']
Chemical potentials change from p1 to p2.
Calculate gq for defect in each charge state.
Calculate Nsites for P_Zn1: 1.245078e+22 cm^-3.
Calculate Nsites for Zn_P1: 2.490156e+22 cm^-3.
Calculate Nsites for Ge_P1: 2.490156e+22 cm^-3.
Calculate Nsites for P_Ge1: 1.245078e+22 cm^-3.
Calculate Nsites for P_i-3: 1.660104e+22 cm^-3.
Calculate Nsites for P_i-1: 1.660104e+22 cm^-3.
...
############ Collecting information from DEC ############
DefectParams.txt 文件:1300 300
0.360000 0.540000
2.067143
P_Zn1 1.245078e+22 2 1.0959 1 0.7854 2 0.602 1 0.3478 2 1.3758 1 1.8193 2 1.8431 1 2.0373 2 3.993075 2.591475
Zn_P1 2.490156e+22 2 0.9803 1 0.4186 2 0.1979 1 x x 1.3004 1 1.6611 2 1.8974 1 2.0551 2 2.013776 3.415376
Ge_P1 2.490156e+22 2 0.017 1 x x x x x x 0.6089 1 1.4605 2 1.7595 1 x x 1.214661 1.681861
P_Ge1 1.245078e+22 2 1.5928 1 0.6776 2 0.3533 1 x x 1.8788 1 2.0787 2 x x x x 2.242467 1.775267
P_i-3 1.660104e+22 2 0.463 1 0.1405 2 0.0186 1 x x 1.1848 1 x x x x x x 3.841352 3.374152
P_i-1 1.660104e+22 2 1.3134 1 0.5638 2 0.2947 1 0.112 2 1.09 1 x x x x x x 4.262836 3.795636
...
生长温度下自洽计算:
DDC模块在T=1300 K的时候计算其缺陷浓度和载流子浓度,并根据电中性条件自洽求解费米能级。
############ First-time self-consistent calculation ############
Fermi level at growth temperature of 1300.000000 K
Fermi level = 0.622739 eV
Fermi level = 0.626234 eV
Fermi level = 0.629768 eV
Fermi level = 0.633339 eV
Fermi level = 0.636946 eV
...
The defect density for one single defect is fixed at the value calculated at T=1300.000000 K.
############ First-time self-consistent calculation ############
工作温度下自洽计算:
DDC模块在T=300 K的时候重新分布每个缺陷各价态的浓度,并根据电中性条件再次自洽求解费米能级。
############ Second-time self-consistent calculation ############
Defect densities in each charge state are redistributed.
Fermi level at working temperature of 300.000000 K
Fermi level = 0.575485 eV
Fermi level = 0.577322 eV
Fermi level = 0.579134 eV
Fermi level = 0.580921 eV
Fermi level = 0.582682 eV
...
############ Second-time self-consistent calculation ############
输出缺陷浓度:
DDC模块在ZnGeP2/ddc目录下,输出三个数据文件与一个图片文件: Fermi.dat Carrier.dat Defect_charge.dat 以及 density.png 。
Output Fermi level as [Fermi.dat].
Output Carrier density as [Carrier.dat].
Output Defect density as [Defect_charge.dat].
############ DDC calculation is done. ############
可使用Origin根据上述数据文件画图,可参考本程序自动输出的图像 density.png ,其中从化学势点p1至p2所得到的缺陷浓度变化图像如下所示:
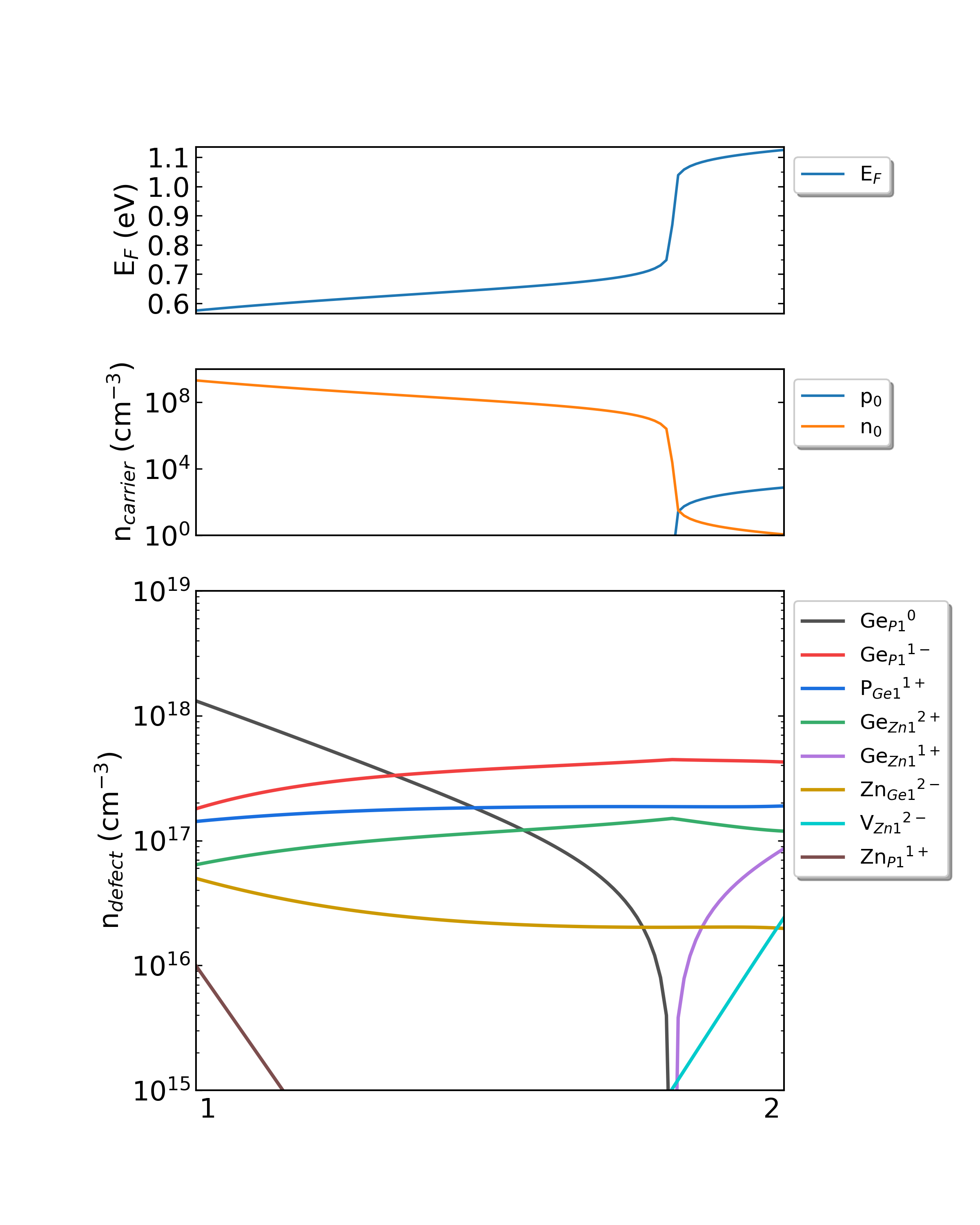
ZnGeP2的化学势从1过渡到2时费米能级、载流子浓度和缺陷浓度。(生长温度:1300K,工作温度:300K)
用户可在 dasp.in 文件中调整生长温度与工作温度,获得不同情况下的缺陷浓度。例如在 dasp.in 文件中按如下修改参数,再次使用命令 dasp 4 执行DDC模块,可以获得如下缺陷浓度变化图像
############## DDC Module ##############
ddc_temperature = 800 300
ddc_mass = 0.36 0.54
ddc_path = 1 2
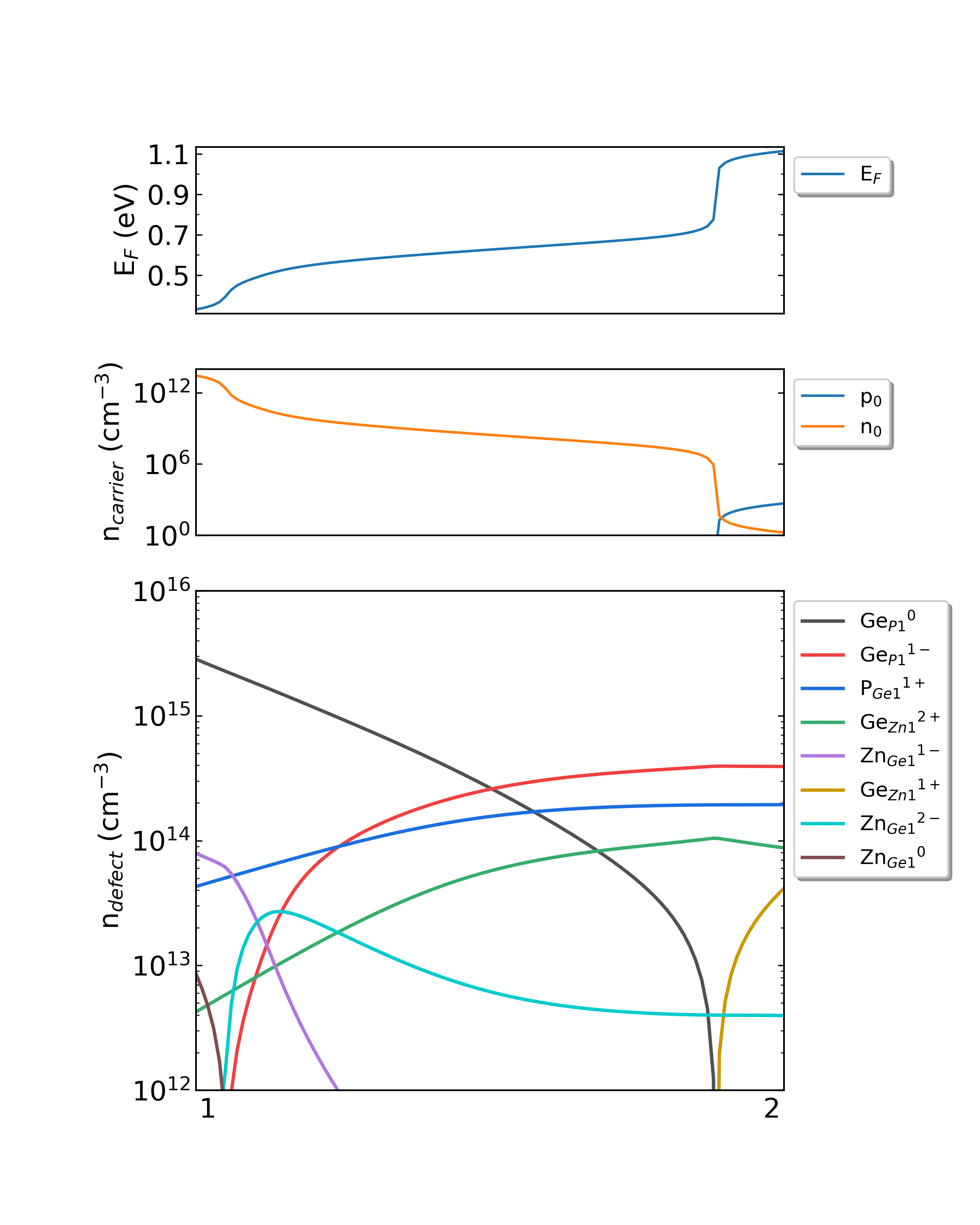
ZnGeP2的化学势从1过渡到2时费米能级、载流子浓度和缺陷浓度。(生长温度:800K,工作温度:300K)
或
############## DDC Module ##############
ddc_temperature = 300 300
ddc_mass = 0.36 0.54
ddc_path = 1 2
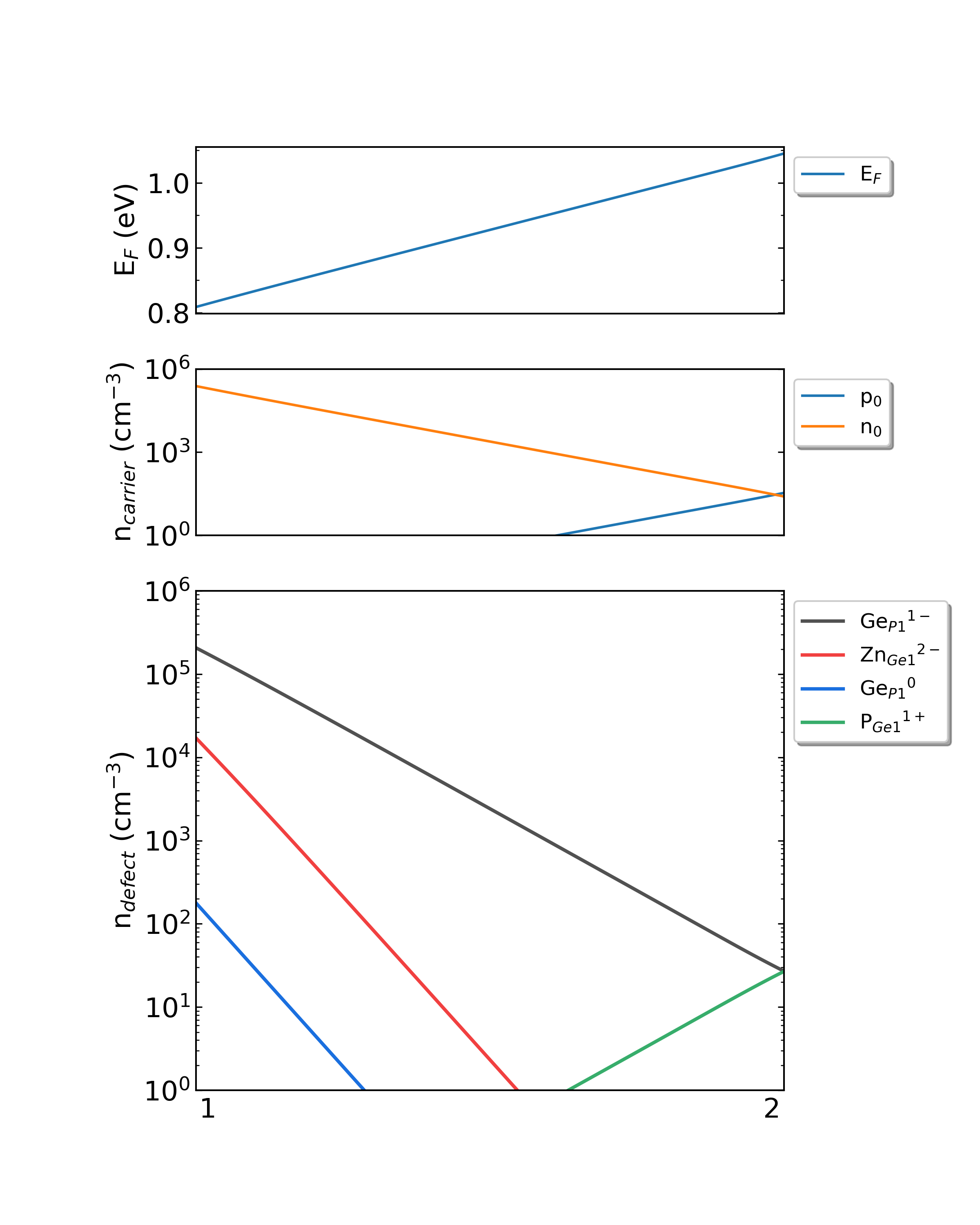
ZnGeP2的化学势从1过渡到2时费米能级、载流子浓度和缺陷浓度。(生长温度:300K,工作温度:300K)
5.4.5. 辐射跃迁系数和光致发光谱计算CDC¶
5.4.5.1. 运行CDC模块¶
############## CDC Module ###############
cdc_defect = Ge_Zn1
cdc_job = pl
cdc_charge = 0 1 # 俘获前的带电状态在前,俘获后的带电状态在后
cdc_band = 864 865 # 带边的序号在前,缺陷能级的序号在后
cdc_temperature = 300
spin_channel = 2
refractive_index = 2.38
dasp 5 执行CDC模块。等待期间无需额外操作。5.4.5.2. 前期计算¶
5cdc.out :----------------------- relaxation calc of initial state -----------------------
finished : /data/home/.../ZnGeP2/cdc/Ge_Zn1/Radiate_calc/_q0_to_q1_/initial_state/relaxation
----------------------- relaxation calc of final state -----------------------
finished : /data/home/.../ZnGeP2/cdc/Ge_Zn1/Radiate_calc/_q0_to_q1_/final_state/relaxation
------------------------------------------------
5cdc.out :Ge_Zn1 : from q0 state to q1 state
hole : from 864 band to 865 band
----------------------- static calc of initial state for transition matrix -----------------------
finished : /data/home/.../ZnGeP2/cdc/Ge_Zn1/Radiate_calc/_q0_to_q1_/initial_state/static_optic
----------------------- static calc of intermediate state for relaxation energy -----------------------
finished : /data/home/.../ZnGeP2/cdc/Ge_Zn1/Radiate_calc/_q0_to_q1_/intermediate_state/static
5cdc.out :transition level is 1.4203 eV
E_zpl ( Energy of zero phonon line ) is 1.4203 eV
total energy of the final state with the initial state configuration is -1142.4152 eV
total energy of the final state is -1142.6177 eV
E_rel (the lattice relaxation energy) is 0.2025 eV
E_emission (the emission energy) is 1.2178 eV
5.4.5.3. 辐射俘获系数计算流程¶
5cdc.out :Radiative carrier capture coefficient is 0.9106*1e-13 cm^3/s
----------------------- End of Calculation for Radiative Capture Rate -----------------------
5.4.5.4. CDC模块PL谱计算流程¶
获取上述数据后,CDC模块会分析HSE泛函优化后缺陷的初态、末态两个结构在广义坐标下的差异 \(ΔQ\) ,并沿着该方向线性地产生一系列结构。
在目录/cdc/Ge_Zn1/Radiate_calc/_q0_to_q1_/final_state与目录/cdc/Ge_Zn1/Radiate_calc/_q0_to_q1_/initial_state中均会出现以下多个静态计算的目录
Q0 Q10 Q-10 Q2 Q-2 Q4 Q-4 Q6 Q-6 Q8 Q-8
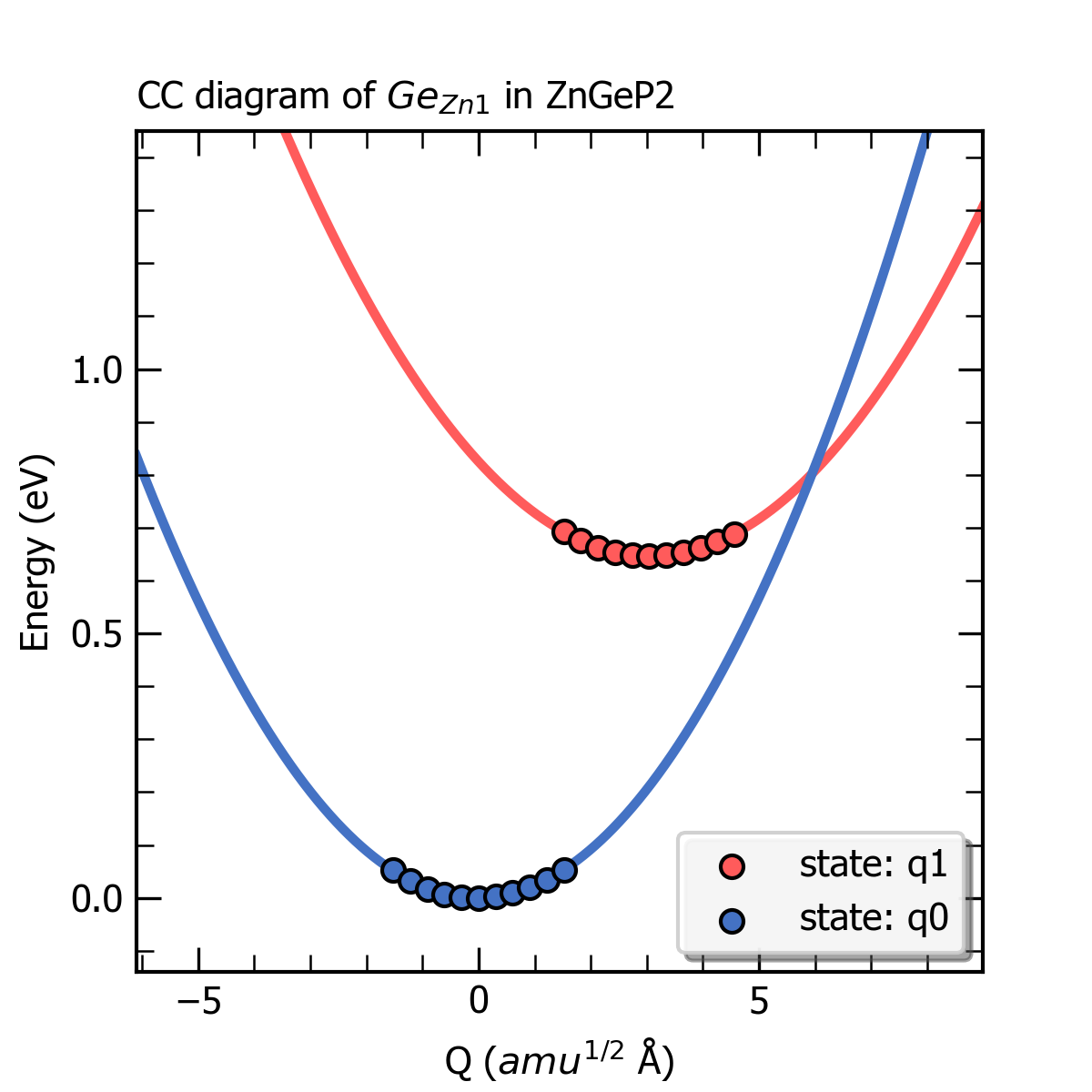
完成上述计算后,CDC模块可以根据产生的结构及对应的缺陷形成能大小得到初态与末态下的有效声子能量、声子波函数以及末态的黄-里斯因子(Huang-Rhys factor),还可以获得该缺陷初态与末态的一维位形图,输出为图片 ccdiagram.png ,如下所示。
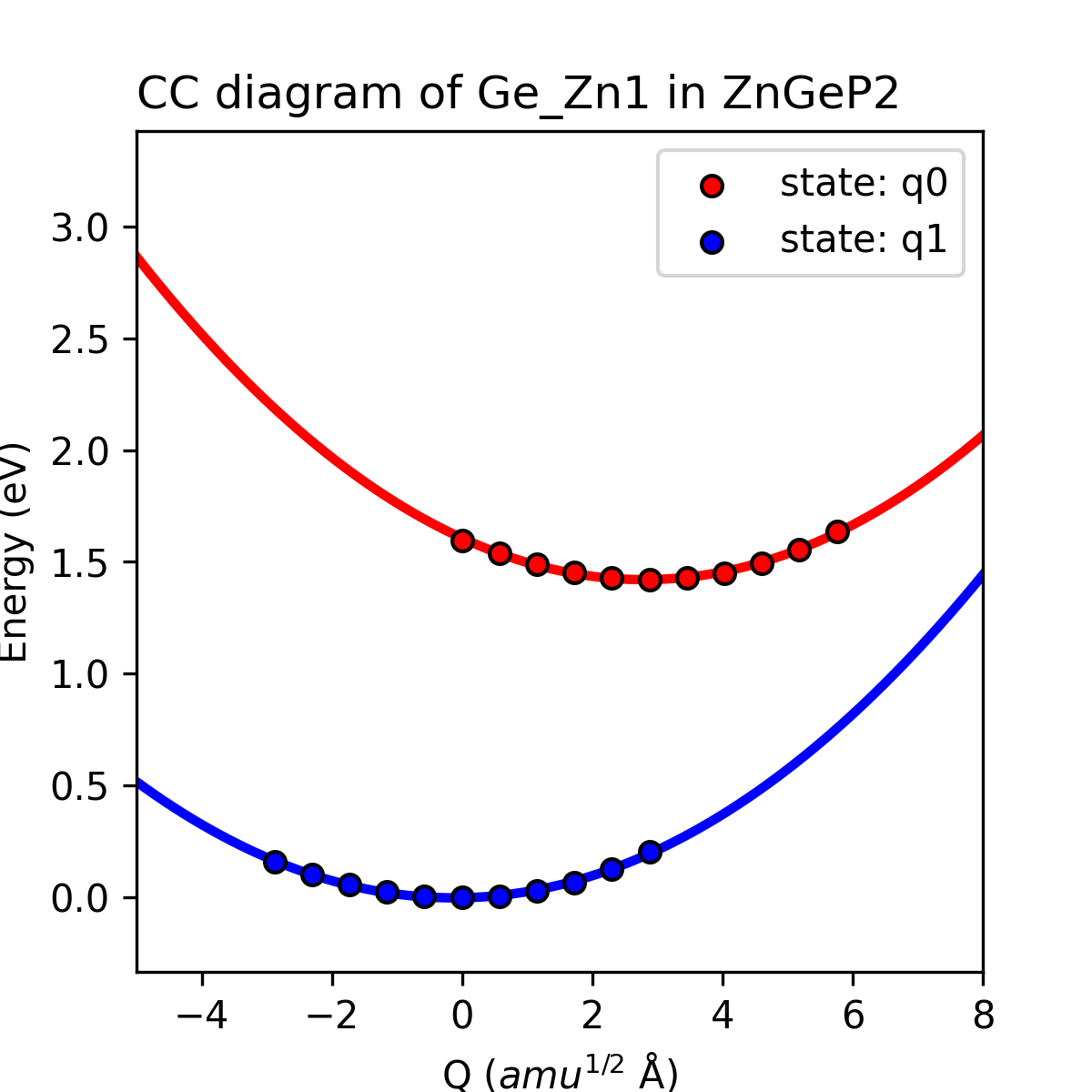
ZnGeP2中缺陷Ge_Zn1的一维位形图。
5cdc.out :----------------------- Calculation for PL Spectrum Start -----------------------
Analysing deltaQ (the structure difference in generalized coordinate) ...
deltaQ between two structures in a.u.:232.2316
deltaQ between two structures in amu^1/2*Angs: 2.879
------------------------------------------------
Generating structures...
...
------------------------------------------------
calculation of initial state and final state all finished
------------------------------------------------
analysing for pl spectrum...
effective phonon energy of the initial state is 0.01397 eV
effective phonon energy of the final state is 0.01338 eV
Huang-Rhys factor of the final state is 15.13453
ccdiagram.png saved in dir /data/home/Zn_Ge_P/cdc/Ge_Zn1/Radiate_calc/_q0_to_q1_
最后,输出PL谱的原始数据 lineshape.dat 以及其尖峰位置、半峰宽,最后图形将存于 lineshape.dat 文件中。
lineshape.dat saved in dir /data/home/test/Zn_Ge_P/CDC_test_0310/cdc/Ge_Zn1/Radiate_calc/_q0_to_q1_
Position of the peak in the lineshape is 1.26 eV
Full width at half maxima of the lineshape is 0.21 eV
analysis for pl spectrum finished
lineshape.png saved in dir /data/home/Zn_Ge_P//cdc/Ge_Zn1/Radiate_calc/_q0_to_q1_
----------------------- End of Calculation for PL Spectrum -----------------------
lineshape.dat 文件如下图所示
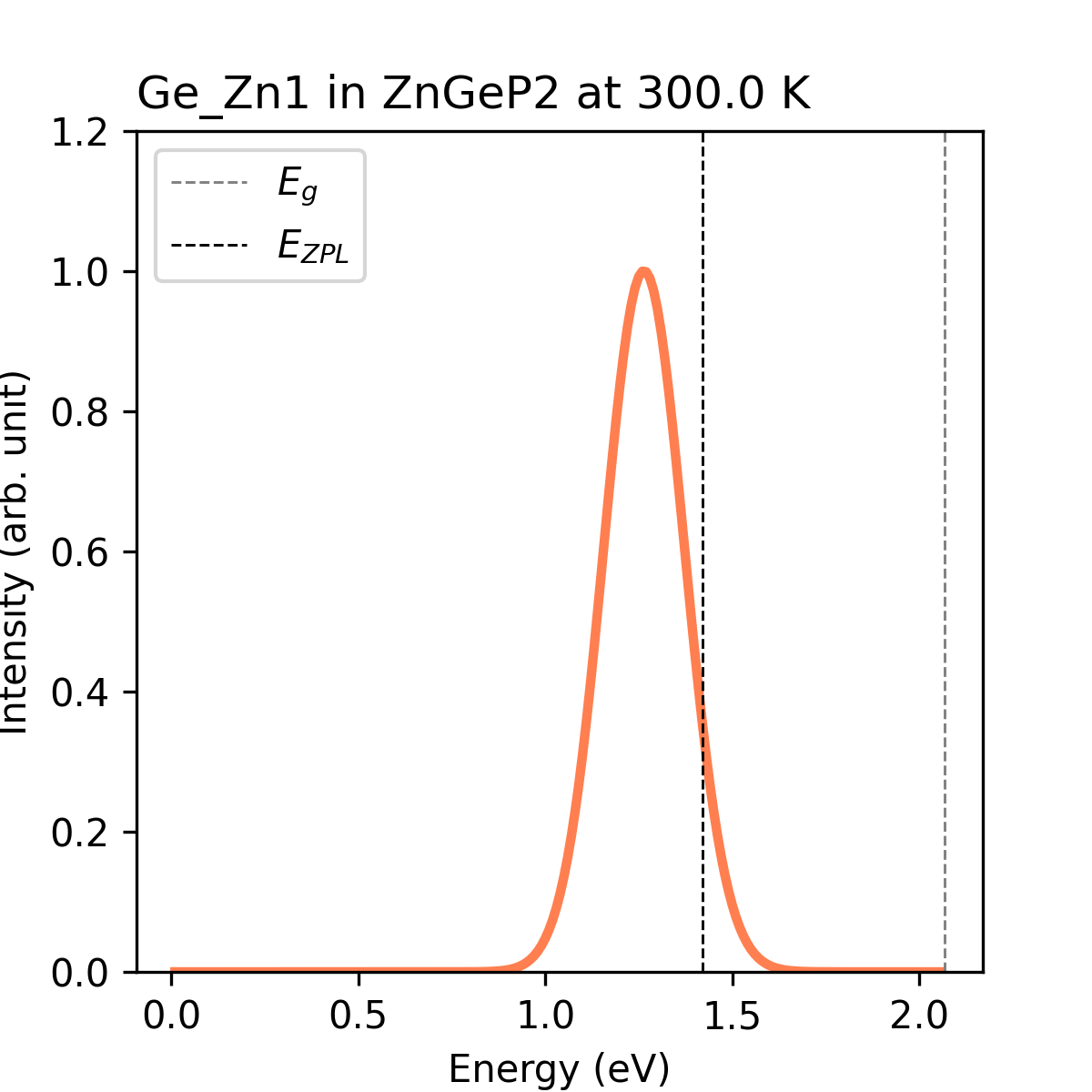
指定温度下ZnGeP2中缺陷Ge_Zn1的PL谱。
5.4.6. 非辐射俘获系数计算CDC¶
5.4.6.1. 运行CDC模块¶
dasp.in 中写入以下信息:############## CDC Module ###############
cdc_defect = Ge_Zn1
cdc_job = nonrad_rate
cdc_charge = +1 0 # 俘获前的带电状态在前,俘获后的带电状态在后
cdc_band = 866 865 # 带边的序号在前,缺陷能级的序号在后
cdc_temperature = 300
spin_channel = 2
dasp 5 执行CDC模块。等待期间无需额外操作。5.4.6.2. 前期计算¶
dasp.in 中 level 的值判断目标缺陷的初态与末态结构是否已完成了HSE泛函下的结构优化。若 level 为2,CDC会额外对目标缺陷做HSE泛函下的结构优化,若 level 为3则跳过结构优化步骤,若 level 为1则会退出该模块的计算。5cdc.out :----------------------- relaxation calc of initial state -----------------------
Finished : /data2/home/.../ZnGeP2/cdc/Ge_Zn1/Nonrad_calc/_q1_to_q0_/initial_state/relaxation
----------------------- relaxation calc of final state -----------------------
Finished : /data2/home/.../ZnGeP2/cdc/Ge_Zn1/Nonrad_calc/_q1_to_q0_/final_state/relaxation
------------------------------------------------
5.4.6.3. CDC模块非辐射俘获系数计算流程¶
完成前期计算得到HSE泛函优化的初态和末态结构后,CDC模块会分析两个结构在广义坐标下的差异 \(ΔQ\) ,并沿着该方向线性地产生一系列结构。
在目录/cdc/Ge_Zn1/Nonrad_calc/_q1_to_q0_/final_state与目录/cdc/Ge_Zn1/Nonrad_calc/_q1_to_q0_/initial_state中均会出现以下多个静态计算的目录:
Q0 Q1 Q-1 Q2 Q-2 Q3 Q-3 Q4 Q-4 Q5 Q-5
此外,在目录/cdc/Ge_Zn1/Nonrad_calc/_q1_to_q0_/final_state/el_ph中产生以下多个用于计算电声耦合常数的静态计算目录:
Q0.0 Q0.49 Q-0.49 Q0.74 Q-0.74
完成上述计算后,CDC模块可以根据产生的结构及对应的缺陷形成能大小得到初态与末态下的有效声子能量、声子波函数以及末态的黄-里斯因子(Huang-Rhys factor)以及用于估算非辐射俘获系数的 \(δ\) 函数的高斯展宽(gaussian smearing)和索莫菲因子(sommerfeld factor),还可以获得该缺陷初态与末态的一维位形图,输出为图片 ccdiagram.png ,如下所示。
ZnGeP2中缺陷Ge_Zn1从+1价到0价的一维位形图。
最后,CDC模块会根据超胞体积、载流子有效质量等数据结合非辐射俘获系数的公式计算输出室温下该系数的大小,并在目录 /cdc 下输出 nonradiative_rate.dat 文件和图片 coefficient.png ,如下所示,其中给出了非辐射俘获系数以及非辐射俘获截面随温度的变化。
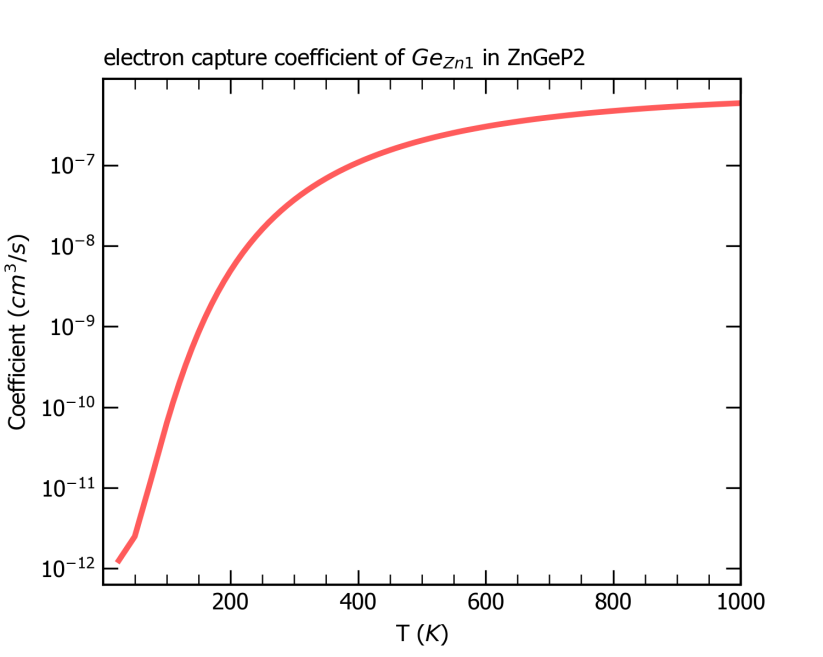
ZnGeP2中缺陷Ge_Zn1从+1价到0价电子俘获系数随温度的变化关系。
5cdc.out :Analysing deltaQ (the structure difference in generalized coordinate) ...
deltaQ between two structures in a.u.:245.3062
deltaQ between two structures in amu^1/2*Angs: 3.0411
------------------------------------------------
Generating structures...
transition level is 1.4203 eV
band gap is 2.0672 eV
E_zpl ( Energy of zero phonon line ) is 0.6469000000000003 eV
Analysing deltaQ (the structure difference in generalized coordinate) ...
deltaQ between two structures in a.u.:245.3062
deltaQ between two structures in amu^1/2*Angs: 3.0411
------------------------------------------------
Generating structures...
------------------------------------------------
...
------------------------------------------------
------------------------------------------------
calculation of initial state and final state all finished
------------------------------------------------
analysing for nonradiative carrier capture coefficient...
electron-phonon coupling constant in eV/(Angs*amu^1/2): 0.02999
barrier for the nonradiative process is 0.15573 eV
try to plot the ccdiagram...
ccdiagram.png saved in dir /data2/home/.../cdc/Ge_Zn1/Nonrad_calc/_q1_to_q0_
initial state phonon energy and wavefunction saved in dir /data2/home/.../cdc/Ge_Zn1/Nonrad_calc/_q1_to_q0_/initial_state/phonon
final state phonon energy and wavefunction saved in dir /data2/home/.../cdc/Ge_Zn1/Nonrad_calc/_q1_to_q0_/final_state/phonon
effective phonon energy of the final state is 0.01373 eV
Huang-Rhys factor of the final state is 15.38015
effective phonon energy of the initial state is 0.01255 eV
gaussian smearing is 0.00941 eV. The sommerfeld factor at 300.0 (K) is 7.936136753462156 .
The carrier capture coefficient at 300.0 (K) is 3.717573134196475e-08 cm^3/s.
see file nonradiative_rate.dat in dir /data2/home/.../cdc for more details.
coefficient.png saved in dir /data2/home/.../cdc/Ge_Zn1/Nonrad_calc/_q1_to_q0_
--------------------- End of Calculation for Nonradiative Capture Rate ---------------------
根据DDC模块得到缺陷和载流子浓度,以及CDC模块得到的非辐射俘获系数,我们可以根据SRH(Shockley–Read–Hall)公式估算出载流子的非辐射俘获寿命 \(τ_{SRH}\)。
dasp.in 文件如下:############## CDC Module ###############
cdc_defect = Ge_Zn1
cdc_job = nonrad_rate
cdc_charge = +1 0 # 俘获前的带电状态在前,俘获后的带电状态在后
cdc_band = 866 865 # 带边的序号在前,缺陷能级的序号在后
cdc_temperature = 300
spin_channel = 2
fitting_method = spline
dasp 5 执行CDC模块。等待期间无需额外操作。CDC模块会分析HSE泛函优化的初态和末态两个结构在广义坐标下的差异 \(ΔQ\) ,并沿着该方向线性地在更大的范围内产生一系列结构。
在目录/cdc/Ge_Zn1/Nonrad_calc/_q1_to_q0_/final_state与目录/cdc/Ge_Zn1/Nonrad_calc/_q1_to_q0_/initial_state中均会出现以下多个静态计算的目录:
Q0 Q1 Q-1 Q2 Q-2 Q3 Q-3 Q4 Q-4 Q5 Q-5
此外,在目录/cdc/Ge_Zn1/Nonrad_calc/_q1_to_q0_/final_state/el_ph中产生以下多个用于计算电声耦合常数的静态计算目录:
Q0.0 Q0.49 Q-0.49 Q0.74 Q-0.74
完成上述计算后,CDC模块可以根据产生的结构及对应的缺陷形成能大小得到初态与末态下的有效声子能量、声子波函数以及末态的黄-里斯因子(Huang-Rhys factor)以及用于估算非辐射俘获系数的 \(δ\) 函数的高斯展宽(gaussian smearing)和索莫菲因子(sommerfeld factor),还可以获得该缺陷初态与末态的一维位形图,输出为图片 ccdiagram.png ,如下所示。
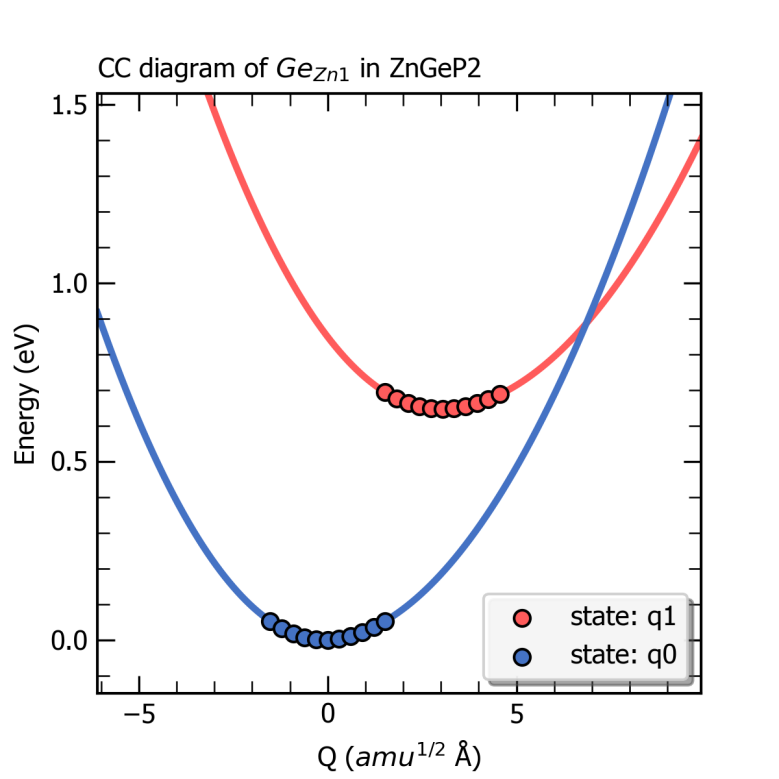
ZnGeP2中缺陷Ge_Zn1从+1价到0价的一维位形图。
可以看到,相比parabolic方法,采用spline的拟合方法将使得俘获势垒增加将近0.1 eV。我们建议总是优先采用parabolic方法进行势能面拟合。
最后,CDC模块会根据超胞体积、载流子有效质量等数据结合非辐射俘获系数的公式计算输出室温下该系数的大小,并在目录 /cdc 下输出 nonradiative_rate.dat 文件和图片 coefficient.png ,如下所示,其中给出了非辐射俘获系数以及非辐射俘获截面随温度的变化。
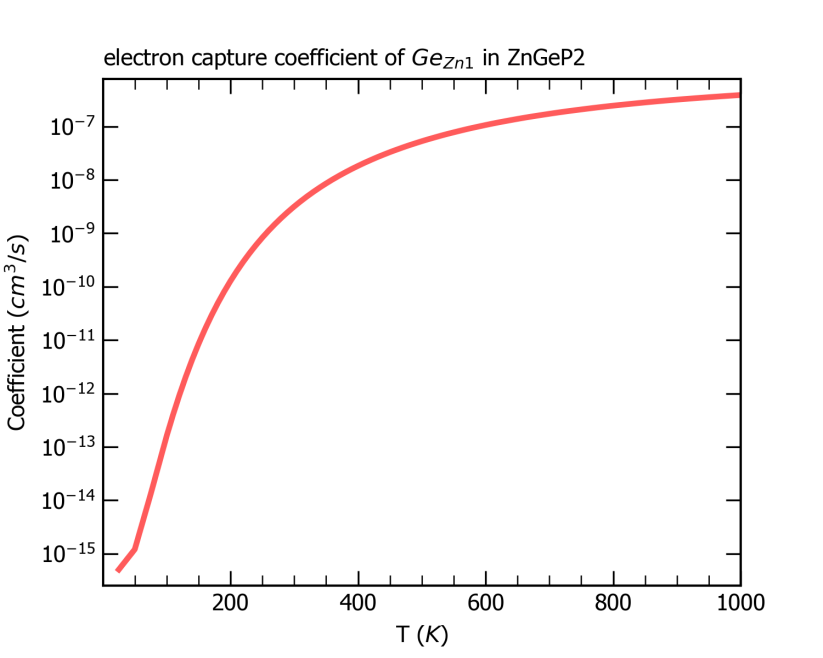
ZnGeP2中缺陷Ge_Zn1从+1价到0价电子俘获系数随温度的变化关系。
5cdc.out :Analysing deltaQ (the structure difference in generalized coordinate) ...
deltaQ between two structures in a.u.:245.3062
deltaQ between two structures in amu^1/2*Angs: 3.0411
-----------------------------------------------
Generating structures...
transition level is 1.4203 eV
band gap is 2.0672 eV
E_zpl ( Energy of zero phonon line ) is 0.6469000000000003 eV
Analysing deltaQ (the structure difference in generalized coordinate) ...
deltaQ between two structures in a.u.:245.3062
deltaQ between two structures in amu^1/2*Angs: 3.0411
-----------------------------------------------
Generating structures...
-----------------------------------------------
..
-----------------------------------------------
-----------------------------------------------
calculation of initial state and final state all finished
-----------------------------------------------
analysing for nonradiative carrier capture coefficient...
electron-phonon coupling constant in eV/(Angs*amu^1/2): 0.02999
barrier for the nonradiative process is 0.23881 eV
try to plot the ccdiagram...
ccdiagram.png saved in dir /data2/home/.../cdc/Ge_Zn1/Nonrad_calc/_q1_to_q0_
initial state phonon energy and wavefunction saved in dir /data2/home/.../cdc/Ge_Zn1/Nonrad_calc/_q1_to_q0_/initial_state/phonon
final state phonon energy and wavefunction saved in dir /data2/home/.../cdc/Ge_Zn1/Nonrad_calc/_q1_to_q0_/final_state/phonon
effective phonon energy of the final state is 0.01338 eV
Huang-Rhys factor of the final state is 14.11924
effective phonon energy of the initial state is 0.01259 eV
gaussian smearing is 0.00945 eV. The sommerfeld factor at 300.0 (K) is 7.93614 .
The carrier capture coefficient at 300.0 (K) is 3.18217e-9 cm^3/s.
see file nonradiative_rate.dat in dir /data2/home/.../cdc for more details.
coefficient.png saved in dir /data2/home/.../cdc/Ge_Zn1/Nonrad_calc/_q1_to_q0_
--------------------- End of Calculation for Nonradiative Capture Rate ---------------------
5.5. 双钙钛矿材料稳定性的快速计算预测¶
在以上的案例中,我们展示了TSC模块可以计算元素化学势,用于DEC模块的缺陷形成能计算。此外,TSC也可以独立运行来分析目标化合物的稳定性,而无需先行做PREPARE模块的计算。
在 dasp.in 中设置 tsc_only = T ,可以直接运行TSC模块,待TSC结束第一阶段分析后,将自行做 level = 1 的第二阶段分析。
以下示例为 Cs2AgBiCl6 与 Rb2LiInI、 K2LiYF6 三种双钙钛矿材料的分析过程。
5.5.1. Cs2AgBiCl6 (预测结果:稳定)¶
5.5.1.1. 准备文件¶
利用 TSC 模块快速分析材料稳定性的第一步仍然是准备好 POSCAR 与 dasp.in 文件。
材料 Cs2AgBiCl6 的 POSCAR 文件可参考 Materials Project 数据库获得,需用户自行优化或设置晶体结构。本案例采取的 POSCAR 文件如下:
Cs2 Ag1 Bi1 Cl6
1.00000000000000
7.7438184481880610 0.0000000000000355 0.0000000000000251
3.8719092240440918 6.7063434983622878 -0.0000000000000092
3.8719092240440918 2.2354478328207339 6.3228012862560394
Cs Ag Bi Cl
2 1 1 6
Direct
0.7500000000000000 0.7500000000000000 0.7500000000000000
0.2500000000000000 0.2500000000000000 0.2500000000000000
0.5000000000000000 0.5000000000000000 0.5000000000000000
-0.0000000000000000 -0.0000000000000000 0.0000000000000000
0.7508700137251050 0.2491299562748926 0.2491299562748926
0.2491299562748926 0.2491299562748926 0.7508700137251050
0.2491299562748926 0.7508700137251050 0.7508700137251050
0.2491299562748926 0.7508700137251050 0.2491299562748926
0.7508700137251050 0.2491299562748926 0.7508700137251050
0.7508700137251050 0.7508700137251050 0.2491299562748926
在 dasp.in 文件中,用户需根据自身情况设置任务脚本相关参数,并设置 tsc_only = T 以及 database_api 。
############## Job Scheduling ##############
cluster = SLURM # (job scheduling system)
node_number = 4 # (number of node)
core_per_node = 32 # (core per node)
queue = normal # (name of queue/partition)
max_time = 24:00:00 # (maximum time for a single DFT calculation)
vasp_path_tsc = /opt/vasp.5.4.4/bin/vasp_std
job_name = submit_job # (name of script)
potcar_path = /opt/POT/potpaw_PBE # (path of pseudopotentials)
max_job = 5
############## TSC Module ##############
database_api = ******************* # (str-list type)
tsc_only = T
plot_3d = T
database_api = ******************* # (str-list type)
# 用于访问Materials Project数据库
tsc_only = T
# 仅进行 level = 1 的稳定性快速分析。
plot_3d = T
# 对四元化合物,本模块可输出三维相图(仅供用户参考),该参数默认为F,设置为T即可输出三维相图。
5.5.1.2. 计算与分析¶
host结构的总能计算(与MP参数保持一致):
TSC模块将使用 Materials Project 数据库提供的输入参数(INCAR,KPOINTS,POTCAR)对用户给定的原胞做结构优化和静态计算,该计算得到的总能与MP数据库的总能是可比的。此步骤是为了得到影响Cs2AgBiCl6稳定性的 关键杂相 。通过目录可以看到:
cd tsc
cd Cs2AgBiCl6/
ls
relaxation1 relaxation2 static
从Cs2AgBiCl6/tsc/2tsc.out中也可以看到程序的运行日志,即产生输入文件、relaxation1、relaxation2、static、数据提取等步骤。
稳定性与关键杂相快速分析:
TSC模块将搜寻MP数据库上所有与Cs2AgBiCl6相竞争的杂项,根据本步骤的输出文件 materials_info.yaml 可以发现,所有考虑到的杂项包括:
secondary_phases:
- - Cs
- Ag
- Bi
- Cl2
- Ag3Bi
- Ag2Cl3
- Ag3Cl
- AgCl
- Cs2AgCl3
- CsAgCl2
- CsAgCl3
- Bi6Cl7
- BiCl3
- Cs3BiCl6
- Cs3Bi2Cl9
- CsBi
- Cs3Bi2
- CsBi2
- Cs3Bi
- CsCl
通过DFT计算的Cs2AgBiCl6的总能与MP数据库中杂相的总能,判断出Cs2AgBiCl6是 稳定的 。
随后,程序将计算获取影响Cs2AgBiCl6稳定性最关键的杂相,本例中包括Ag、Cs、Bi、Cl2、AgCl、CsAgCl2、Cs3BiCl6、CsAgCl3、Cl2Cs3Bi2Cl9 。在 2tsc.out 中可看到相关的信息:
...
analysing the thermodynamic stability of Cs2AgBiCl6.
The stability of Cs2AgBiCl6 is: True.
key phases of Cs2AgBiCl6 are: AgCl Ag Cs3Bi2Cl9 CsAgCl2 Cs3BiCl6 CsAgCl3 Cs Bi Cl2 .
analysing of Cs2AgBiCl6 is done.
...
同时, materials_info.yaml 文件中也有此信息输出:
key_phases:
- AgCl
- CsAgCl2
- CsAgCl3
- Ag
- Cs3BiCl6
- Cs3Bi2Cl9
- Cs
- Bi
- Cl2
host与杂相结构的总能计算:
由于该目标化合物是稳定的,因此在确定关键杂相后,TSC模块还做第二阶段的分析与计算,即将根据用户提供的赝势路径中的POTCAR文件,按 level=1 计算Cs2AgBiCl6及以上各类关键杂相的总能。 2tsc.out 如下:
...
Job ******** submitted: /home/test/Cs2AgBiCl6/tsc/Cs2AgBiCl6/static_recalc
Job ******** submitted: /home/test/Cs2AgBiCl6/tsc/AgCl/static_recalc
Job ******** submitted: /home/test/Cs2AgBiCl6/tsc/CsAgCl2/static_recalc
...
Succeed job 12267.host2: /home/test/Cs2AgBiCl6/tsc/Cs2AgBiCl6/static_recalc
Succeed job 12269.host2: /home/test/Cs2AgBiCl6/tsc/AgCl/static_recalc
Succeed job 12271.host2: /home/test/Cs2AgBiCl6/tsc/CsAgCl2/static_recalc
...
若有发现存在任务计算出错,可自行修改第一性计算所需相关参数,如 INCAR 或 KPOINTS 等文件内容,然后再次运行具体可参考常见问题板块。
化学势的计算:
根据DFT计算的总能,计算Cs2AgBiCl6的形成能和化学势稳定区间,TSC模块给出8个化学势的端点值,写入 dasp.in :
# 顺序与POSCAR中元素顺序一致,即第一列是Cs,第二列是Ag,第三列是Bi,第四列是Cl。
E_pure = -0.836 -2.6987 -3.8871 -1.7877
p1 = -3.5853 -0.4806 -2.9502 -0.5666
p2 = -3.1047 -0.0 -1.5084 -1.0472
p3 = -4.1861 -0.781 -3.2506 -0.2662
p4 = -3.4051 -0.0 -0.9076 -1.0472
p5 = -3.5373 -0.5286 -2.9982 -0.5666
p6 = -3.0087 0.0 -1.4124 -1.0952
p7 = -3.7897 -0.781 -3.2506 -0.3983
p8 = -3.0087 -0.0 -0.9076 -1.1793
在 2tsc.out 可以看到程序执行完毕的输出:
analysing the thermodynamic stability of Cs2AgBiCl6.
The stability of Cs2AgBiCl6 is: True.
key phases of Cs2AgBiCl6 are: AgCl Ag Cs3Bi2Cl9 CsAgCl2 Cs3BiCl6 CsAgCl3 Cs Bi Cl2 .
analysing of Cs2AgBiCl6 is done.
sub-module of tsc: 'auto thermodynamic calculation' ends successfully.
---------------------------
DASP-TSC finished
对于三元与四元的化合物,TSC模块将输出稳定区域图像,及稳定区域各端点处的化学势。对于二维图像,通过目录可以看到:
cd tsc
cd 2d-figures/
ls
fig-Cs2AgBiCl6.png fig-Cs2AgBiCl6_recalc.png stable_2d.out stable_recalc_2d.out
目录Cs2AgBiCl6/tsc/2d-figures/中的四个文件分别是两次计算与分析过程中绘制的稳定区域图像以及图像中各端点处的化学势。
查看文件 stable_2d.out 与 fig-Cs2AgBiCl6.png 。图 fig-Cs2AgBiCl6.png 的横纵坐标分别是图中所标识元素的化学势,阴影区域则是目标化合物的稳定区域,其边界的每一条线是相应所标识材料恰好处于形成与未形成的临界情况下的化学势曲线,这是第一次计算与分析过程输出的图像。
Bi Ag Cs Cl
------- ------- ------- -------
-2.9709 -0.3807 -3.5994 -0.5224
-3.1086 -0.5184 -3.5994 -0.4765
-2.8656 -0.5184 -3.5994 -0.517
-1.6748 -0.1214 -3.5994 -0.7817
Cs2AgBiCl6的稳定区域图(来自MP数据库)
查看文件 stable_recalc_2d.out 与 fig-Cs2AgBiCl6_recalc.png ,这是第二次计算与分析过程输出的数据与图像。
Bi Ag Cs Cl
------- ------- ------- -------
-2.9562 -0.4866 -3.5974 -0.5606
-3.0583 -0.5887 -3.5974 -0.5265
-2.6737 -0.5887 -3.5974 -0.5906
-1.4845 -0.1923 -3.5974 -0.8549
Cs2AgBiCl6的稳定区域图(来自第二阶段计算)
对于三维图像,通过目录可以看到:
cd tsc
cd 3d-figures/
ls
fig-Cs2AgBiCl6_3d.png fig-Cs2AgBiCl6_3d_recalc.png stable.out stable_recalc.out
目录Cs2AgBiCl6/tsc/3d-figures/中的四个文件分别是两次计算与分析过程中绘制的稳定区域图像以及图像中各端点处的化学势。
查看文件 stable.out 与 fig-Cs2AgBiCl6_3d.png 。图 fig-Cs2AgBiCl6_3d.png 中所标识三种元素的化学势构成三个坐标轴,红色线条包围区域为Cs2AgBiCl6的三维稳定区域,这是第一次计算与分析过程输出的图像,该区域各点坐标可通过文件 stable.out 获取。
Cs Ag Bi Cl
-------- ------------ -------- ---------
-3.17837 6.73289e-17 -1.90967 -0.903106
-3.47796 -0 -1.31049 -0.903106
-3.08102 -3.47289e-18 -1.81231 -0.951782
-3.08102 -0 -1.31049 -1.03542
-3.51867 -0.340294 -2.93055 -0.562812
-4.11785 -0.639883 -3.23014 -0.263223
-3.46999 -0.38897 -2.97922 -0.562812
-3.7209 -0.639883 -3.23014 -0.395537
Cs2AgBiCl6的三维稳定区域图(来自MP数据库)
查看文件 stable_recalc_3d.out 与 fig-Cs2AgBiCl6_3d_recalc.png ,这是第二次计算与分析过程输出的数据与图像。
Cs Ag Bi Cl
------- ------------ ------- ---------
-3.5853 -0.4806 -2.9502 -0.5666
-3.1047 -3.63857e-17 -1.5084 -1.0472
-4.1861 -0.781 -3.2506 -0.2662
-3.4051 -0 -0.9076 -1.0472
-3.5373 -0.5286 -2.9982 -0.5666
-3.0087 0 -1.4124 -1.0952
-3.7897 -0.781 -3.2506 -0.398333
-3.0087 -0 -0.9076 -1.17933
Cs2AgBiCl6的三维稳定区域图(来自第二阶段计算)
5.5.2. K2LiYF6 (预测结果:不稳定)¶
5.5.2.1. 准备POSCAR与dasp.in¶
材料 K2LiYF6 的 POSCAR 文件可参考 Materials Project 数据库获得,需用户自行优化或设置晶体结构。本案例采取的 POSCAR 文件及其可视化图形显示如下:
K8 Li4 Y4 F24
1.0
8.557390 0.000000 0.000000
0.000000 8.557390 0.000000
0.000000 0.000000 8.557390
K Li Y F
8 4 4 24
direct
0.250000 0.250000 0.750000 K
0.250000 0.750000 0.750000 K
0.250000 0.750000 0.250000 K
0.250000 0.250000 0.250000 K
0.750000 0.250000 0.250000 K
0.750000 0.750000 0.250000 K
0.750000 0.750000 0.750000 K
0.750000 0.250000 0.750000 K
0.500000 0.000000 0.000000 Li
0.500000 0.500000 0.500000 Li
0.000000 0.000000 0.500000 Li
0.000000 0.500000 0.000000 Li
...
K2LiYF6的晶体结构示意图
在 dasp.in 文件中,用户需根据自身情况设置任务脚本相关参数,并设置 tsc_only = T 以及 database_api 。具体可参考 Cs2AgBiCl6 案例所述。
5.5.2.2. 计算与分析¶
host结构的总能计算(与MP参数保持一致):
TSC模块将使用 Materials Project 数据库提供的输入参数(INCAR,KPOINTS,POTCAR)对用户给定的原胞做结构优化和静态计算,该计算得到的总能与MP数据库的总能是可比的。此步骤是为了得到影响Cs2AgBiCl6稳定性的 关键杂相 。通过目录可以看到:
cd tsc
cd K2LiYF6/
ls
relaxation1 relaxation2 static
从K2LiYF6/tsc/2tsc.out中也可以看到程序的运行日志,即产生输入文件、relaxation1、relaxation2、static、数据提取等步骤。
稳定性与关键杂相快速分析:
TSC模块将搜寻MP数据库上所有与K2LiYF6相竞争的杂项,根据本步骤的输出文件 materials_info.yaml 可以发现,所有考虑到的杂项包括:
secondary_phases:
- - K
- Li
- Y
- F
- KF2
- KF3
- KF
- KF5
- KLiYF5
- K3YF6
- KYF4
- K2YF5
- KY3F10
- KY2F7
- LiF
- LiF3
- LiYF4
- LiYF2
- Li3YF6
- YF3
- K3Li
- KLi3
- K3Y
- LiY3
- Li3Y
通过DFT计算的K2LiYF6的总能与MP数据库中杂相的总能,判断出K2LiYF6是 不稳定的 。在 2tsc.out 中可看到相关的信息:
...
analysing the thermodynamic stability of K2LiYF6.
The stability of K2LiYF6 is: False.
K2LiYF6 may decompose into ['K2YF5', 'LiF'].
you can set tag: 'excluded_phase' to get some reference values of chemical potentials.
analysing of K2LiYF6 is done.
...
K2LiYF6的 energy_above_hull (eV/atom)数值输出在文件 materials_info.yaml 中,其值为正,与“该化合物不稳定”的分析结果相符。
e_above_hull: 0.0466
其分解产物包括K2YF5、LiF,这一点也可从输出文件 materials_info.yaml 获知:
decomp:
- K2YF5
- LiF
由于K2LiYF6不稳定,目前本模块无法判断出关键杂相,也无法输出稳定区域图像,而且,TSC模块不会进一步做第二阶段的计算。
若用户仍然希望做第二阶段计算,获得稳定区域化学势,则可以按照 2tsc.out 文件的提示,可以设置 excluded_phase 来排除某些杂项对目标化合物稳定性的影响,一般选择排除分解物中的一种或多种,来使得目标化合物拥有可参考的稳定区域。即,在 dasp.in 文件中设置 excluded_phase = LiF 或 excluded_phase = K2YF5 ,亦或 excluded_phase = LiF K2YF5 ,然后重新运行 TSC 模块。此时,有可能该化合物仍然不稳定,则可以将当前分解路径中的某些化合物追加写入 excluded_phase ,然后再次运行TSC模块。
5.5.2.3. 追加计算与分析¶
在本例中,排除某些特定杂项后使得目标化合物存在稳定区域的 dasp.in 文件参数如下所示:
############## Job Scheduling ##############
cluster = SLURM # (job scheduling system)
node_number = 4 # (number of node)
core_per_node = 32 # (core per node)
queue = normal # (name of queue/partition)
max_time = 24:00:00 # (maximum time for a single DFT calculation)
vasp_path_tsc = /opt/vasp.5.4.4/bin/vasp_std
job_name = submit_job # (name of script)
potcar_path = /opt/POT/potpaw_PBE # (path of pseudopotentials)
max_job = 5
############## TSC Module ##############
database_api = ******************* # (str-list type)
tsc_only = T
plot_3d = T
excluded_phase = K2LiYF6 K2YF5 LiF KLiYF5 LiYF4
本案例中排除的杂相包括 K2LiYF6,K2YF5,LiF,KLiYF5,LiYF4。
对于不稳定的化合物,由于排除的杂相不同,稳定区域可能不同,上述参数仅供参考。
因为总能计算已经完成,因此很快可以在 2tsc.out 中可看到相关的稳定性分析信息:
analysing the thermodynamic stability of K2LiYF6.
excluded phase of K2LiYF6: K2LiYF6 K2YF5 LiF KLiYF5 LiYF4 .
The stability of K2LiYF6 is: True.
key phases of K2LiYF6 are: KYF4 K3YF6 Li3YF6 KF2 KF Li KY3F10 KY2F7 F2 K Y .
file key_phases_info_recalc.yaml generated.
analysing of K2LiYF6 is done.
并且针对上述信息中提到的关键杂相,第二阶段的计算已经开始, 2tsc.out 如下:
...
Job ******** submitted: /home/fudan/daike/KLiYF/DASP_0219/tsc/K2LiYF6/static_recalc
Job ******** submitted: /home/fudan/daike/KLiYF/DASP_0219/tsc/KYF4/static_recalc
Job ******** submitted: /home/fudan/daike/KLiYF/DASP_0219/tsc/K3YF6/static_recalc
Job ******** submitted: /home/fudan/daike/KLiYF/DASP_0219/tsc/Li3YF6/static_recalc
...
Succeed job ********: /home/fudan/daike/KLiYF/DASP_0219/tsc/K2LiYF6/static_recalc
Succeed job ********: /home/fudan/daike/KLiYF/DASP_0219/tsc/KYF4/static_recalc
Succeed job ********: /home/fudan/daike/KLiYF/DASP_0219/tsc/K3YF6/static_recalc
Succeed job ********: /home/fudan/daike/KLiYF/DASP_0219/tsc/KF2/static_recalc
...
若有发现存在任务计算出错,可自行修改第一性计算所需相关参数,如 INCAR 或 KPOINTS 等文件内容,然后再次运行TSC,具体可参考常见问题板块。
化学势的计算:
根据DFT计算的总能,计算K2LiYF6的形成能和化学势稳定区间,TSC模块给出14个化学势的端点值,写入 dasp.in :
# 顺序与POSCAR中元素顺序一致,即第一列是K,第二列是Li,第三列是Y,第四列是F。
E_pure = -1.086 -1.8579 -6.453 -1.8583
p1 = -0.0059 0.0 -0.1727 -5.5633
p2 = -5.5692 -5.5633 -16.8626 0.0
p3 = -0.4569 0.0 -0.6237 -5.3378
p4 = -5.7947 -5.3378 -16.6371 0.0
p5 = -5.4204 -5.4145 -16.9064 -0.0671
p6 = -5.5546 -5.5487 -16.9064 0.0
p7 = -0.0059 0.0 -0.6629 -5.4816
p8 = -5.4204 -4.1335 -18.1874 -0.0671
p9 = -5.5546 -4.2677 -18.1874 0.0
p10 = -1.2869 0.0 -5.7869 -4.2006
p11 = -1.2869 0.0 -2.0624 -4.8213
p12 = -6.1083 -4.8213 -16.5265 0.0
p13 = -0.5489 0.0 -0.7709 -5.2826
p14 = -5.8315 -5.2826 -16.6187 0.0
在 2tsc.out 可以看到程序执行完毕的输出:
analysing the thermodynamic stability of K2LiYF6.
excluded phase of K2LiYF6: K2LiYF6 K2YF5 LiF KLiYF5 LiYF4 .
The stability of K2LiYF6 is: True.
key phases of K2LiYF6 are: K3YF6 Li3YF6 KF2 KF KYF4 F2 Li KY3F10 KY2F7 K Y .
analysing of K2LiYF6 is done.
sub-module of tsc: 'auto thermodynamic calculation' ends successfully.
---------------------------
DASP-TSC finished
对于三元与四元的化合物,TSC模块将输出稳定区域图像,及稳定区域各端点处的化学势。对于二维图像,通过目录可以看到:
cd tsc
cd 2d-figures/
ls
fig-K2LiYF6.png fig-K2LiYF6_recalc.png stable_2d.out stable_recalc_2d.out
目录K2LiYF6/tsc/2d-figures/中的四个文件分别是两次计算与分析过程中绘制的稳定区域图像以及图像中各端点处的化学势。
查看文件 stable_2d.out 与 fig-K2LiYF6.png 。图 fig-K2LiYF6.png 的横纵坐标分别是图中所标识元素的化学势,阴影区域
则是目标化合物的稳定区域,其边界的每一条线
是相应所标识材料恰好处于形成与未形成的临界情况下的化学势曲线,这是第一次计算与分析过程输出的图像。
Y Li K F
-------- ------- ------- -------
-7.3952 -1.7304 -3.0425 -3.0094
-11.1139 -1.7304 -3.0425 -2.3896
-9.8349 -3.0094 -3.0425 -2.3896
-9.3452 -3.0094 -3.0425 -2.4712
-8.4446 -2.5591 -3.0425 -2.6964
-8.3183 -2.4689 -3.0425 -2.7325
K2LiYF6的稳定区域图(来自MP数据库)
查看文件 stable_recalc_2d.out 与 fig-K2LiYF6_recalc.png ,这是第二次计算与分析过程输出的数据与图像。
Y Li K F
-------- ------- ------- -------
-7.373 -1.7702 -3.0571 -3.0512
-11.0975 -1.7702 -3.0571 -2.4304
-9.8165 -3.0512 -3.0571 -2.4304
-9.3263 -3.0512 -3.0571 -2.5121
-8.4243 -2.6002 -3.0571 -2.7376
-8.2955 -2.5082 -3.0571 -2.7744
K2LiYF6的稳定区域图(来自第二阶段分析)
对于三维图像,通过目录可以看到:
cd tsc
cd 3d-figures/
ls
fig-K2LiYF6_3d.png fig-K2LiYF6_3d_recalc.png stable.out stable_recalc.out
目录K2LiYF6/tsc/3d-figures/中的四个文件分别是两次计算与分析过程中绘制的稳定区域图像以及图像中各端点处的化学势。
查看文件 stable.out 与 fig-K2LiYF6_3d.png 。图 fig-K2LiYF6_3d.png 中所标识三种元素的化学势构成三个坐标轴,红色线条包围区域为K2LiYF6的三维稳定区域,这是第一次计算与分析过程输出的图像,该区域各点坐标可通过文件 stable.out 获取。
K Li Y F
---------- ------------ ---------- ------------
-0.0331078 0 -0.806716 -5.39901
-1.31209 0 -5.92265 -4.12003
-0.0331078 0 -0.316988 -5.48063
-0.483403 0 -0.767283 -5.25549
-0.573574 0 -0.911557 -5.20138
-1.31209 2.75724e-17 -2.20396 -4.73981
-5.49866 -5.46555 -16.8041 5.92119e-16
-5.49866 -4.18657 -18.0831 5.92119e-16
-5.51374 -5.48063 -16.7589 1.77636e-15
-5.73889 -5.25549 -16.5337 1.77636e-15
-5.77496 -5.20138 -16.5157 1.18424e-15
-6.0519 -4.73981 -16.4234 0
-5.36558 -5.33247 -16.8041 -0.0665391
-5.36558 -4.05349 -18.0831 -0.0665391
K2LiYF6的三维稳定区域图(来自MP数据库)
查看文件 stable_recalc_3d.out 与 fig-K2LiYF6_3d_recalc.png ,这是第二次计算与分析过程输出的数据与图像。
K Li Y F
-------- ------------ -------- ------------
-0.0059 0 -0.1727 -5.5633
-5.5692 -5.5633 -16.8626 0
-0.4569 0 -0.6237 -5.3378
-5.7947 -5.3378 -16.6371 0
-5.4204 -5.4145 -16.9064 -0.0671
-5.5546 -5.5487 -16.9064 0
-0.0059 0 -0.6629 -5.4816
-5.4204 -4.1335 -18.1874 -0.0671
-5.5546 -4.2677 -18.1874 5.92119e-16
-1.2869 0 -5.7869 -4.2006
-1.2869 1.56171e-17 -2.0624 -4.82135
-6.10825 -4.82135 -16.5265 0
-0.5489 0 -0.7709 -5.2826
-5.8315 -5.2826 -16.6187 5.92119e-16
K2LiYF6的三维稳定区域图(来自第二阶段计算)
5.5.3. Rb2LiInI6 (预测结果:不稳定)¶
5.5.3.1. 准备POSCAR与dasp.in¶
材料 Rb2LiInI6 的 POSCAR 文件可参考 Materials Project 数据库获得,需用户自行优化或设置晶体结构。本案例采取的 POSCAR 文件如下:
Rb2 Li1 In1 I6
1.0
7.7485766411 0.0000000000 0.0000000000
3.8742883205 6.7104642143 0.0000000000
3.8742883205 2.2368214048 6.3266863345
Rb Li In I
2 1 1 6
Direct
0.750000000 0.750000000 0.750000000
0.250000000 0.250000000 0.250000000
0.500000000 0.500000000 0.500000000
0.000000000 0.000000000 0.000000000
0.750886977 0.249112993 0.249112993
0.249112993 0.249112993 0.750886977
0.249112993 0.750886977 0.750886977
0.249112993 0.750886977 0.249112993
0.750886977 0.249112993 0.750886977
0.750886977 0.750886977 0.249112993
在 dasp.in 文件中,用户需根据自身情况设置任务脚本相关参数,并设置 tsc_only = T 以及 database_api 。具体可参考 Cs2AgBiCl6 案例所述。
5.5.2.2. 计算与分析¶
host结构的总能计算(与MP参数保持一致):
TSC模块将使用 Materials Project 数据库提供的输入参数(INCAR,KPOINTS,POTCAR)对用户给定的原胞做结构优化和静态计算,该计算得到的总能与MP数据库的总能是可比的(参照前文案例)。
稳定性与关键杂相快速分析:
TSC模块将搜寻MP数据库上所有与Rb2LiInI6相竞争的杂项,根据本步骤的输出文件 materials_info.yaml 可以发现,所有考虑到的杂项包括:
secondary_phases:
- - Rb
- Li
- In
- I
- InI4
- InI3
- InI
- InI2
- LiInI4
- Rb3InI6
- RbInI3
- RbInI4
- LiI
- RbI
- RbI3
- LiIn3
- Li3In
- LiIn
- LiIn2
- Li5In4
- Li2In
- Li13In3
- Li3In2
- Rb3In
- RbIn4
- Rb2In3
- Rb8In11
- Rb3Li
通过DFT计算的Rb2LiInI6的总能与MP数据库中杂相的总能,判断出Rb2LiInI6是 不稳定的 。在 2tsc.out 中可看到相关的信息:
...
analysing the thermodynamic stability of Rb2LiInI6.
The stability of Rb2LiInI6 is: False.
Rb2LiInI6 may decomposed into ['RbInI4', 'LiI', 'RbI'].
you can set tag: 'excluded_phase' to get some reference values of chemical potentials.
analysing of Rb2LiInI6 is done.
...
Rb2LiInI6 的 energy_above_hull (eV/atom) 数值输出在文件 materials_info.yaml 中,其值为正,与“该化合物不稳定”的分析结果相符。
e_above_hull: 0.0819
其分解产物包括RbInI4、LiI、RbI,这一点也可从输出文件 materials_info.yaml 获知:
decomp:
- RbInI4
- LiI
- RbI
由于Rb2LiInI6不稳定,目前本模块无法判断出关键杂相,也无法输出稳定区域图像,而且,TSC模块不会进一步做第二阶段的计算。
若用户仍然希望做第二阶段计算,获得稳定区域化学势,则可以按照 2tsc.out 文件的提示,可以设置 excluded_phase 来排除某些杂项对目标化合物稳定性的影响,一般选择排除分解物中的一种或多种,来使得目标化合物拥有可参考的稳定区域。
即,在 dasp.in 文件中设置 excluded_phase = RbInI4 或 excluded_phase = LiI ,亦或excluded_phase = RbInI4 RbI 等 ,然后重新运行 TSC 模块。此时,有可能该化合物仍然不稳定,则可以将当前分解路径中的某些化合物追加写入 excluded_phase ,然后再次运行TSC模块。(可参考K2LiYF6的分析过程)
5.6. GaN中 \(C_N\) 缺陷的非辐射俘获系数计算¶
5.6.1. 运行CDC模块¶
dasp.in 中 level 为3, \(C_N\) 缺陷的初态与末态结构已完成了HSE泛函的结构优化。dasp.in 中写入以下信息:############## CDC Module ###############
cdc_job = nonrad_rate
cdc_defect = C_N1
cdc_charge = -1 0 #俘获前带电状态在前,俘获后带电状态在后
cdc_temperature = 300
cdc_band = 253 256 #带边的序号在前,缺陷能级的序号在后
spin_channel = 2
refractive_index = 2.38
dasp 5 执行CDC模块。等待期间无需额外操作。5.6.2. 非辐射俘获系数计算流程¶
在HSE泛函优化的初态和末态结构基础上,CDC模块会分析两个结构在广义坐标下的差异 \(ΔQ\) ,并沿着该方向线性地产生一系列结构。
在目录 /cdc/C_N1/Nonrad_calc/_q-1_to_q0_/initial_state 与目录 /cdc/C_N1/Nonrad_calc/_q-1_to_q0_/final_state 中均会出现以下多个静态计算的目录:
Q0 Q1 Q-1 Q2 Q-2 Q3 Q-3 Q4 Q-4 Q5 Q-5
此外,在目录 /cdc/C_N1/Nonrad_calc/_q-1_to_q0_/final_state/el_ph 中产生以下多个用于计算电声耦合常数的静态计算目录:
Q0.0 Q-1.1 Q1.1 Q-1.64 Q1.64
完成上述计算后,CDC模块可以根据产生的结构及对应的缺陷形成能大小得到初态与末态下的有效声子能量、声子波函数以及末态的黄-里斯因子(Huang-Rhys factor)以及用于估算非辐射俘获系数的 \(δ\) 函数的高斯展宽(gaussian smearing)和索莫菲因子(sommerfeld factor),还可以获得该缺陷初态与末态的一维位形图,输出为图片 ccdiagram.png ,如下所示。
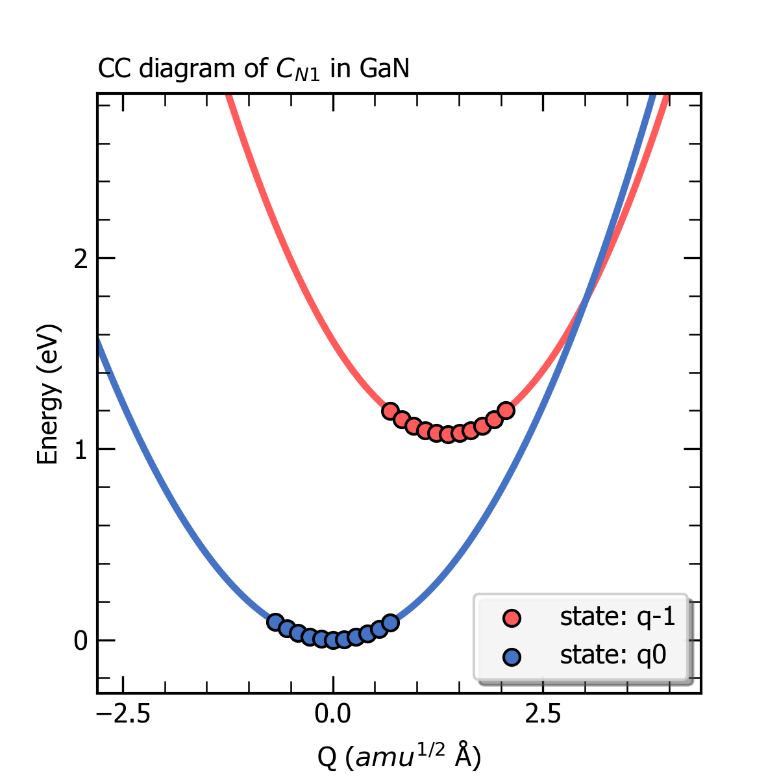
GaN中缺陷C_N1从-1价到0价的一维位形图。
最后,CDC模块会根据超胞体积、载流子有效质量等数据结合非辐射俘获系数的公式计算输出室温下该系数的大小,并在目录 /cdc 下输出 nonradiative_rate.dat 文件和图片 coefficient.png ,如下所示,其中给出了非辐射俘获系数随温度的变化。可以看到,与文献 1 中的Fig. 5基本一致(由于本例中0/-转变能级略大,因此俘获系数略小)。
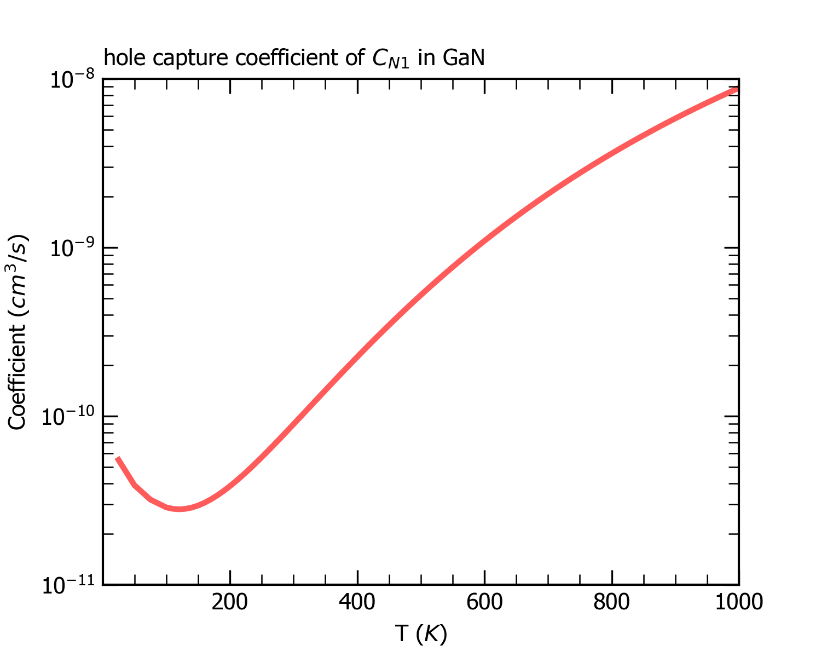
GaN中缺陷C_N1从-1价到0价空穴俘获系数随温度的变化关系。
5cdc.out :Analysing deltaQ (the structure difference in generalized coordinate) ...
deltaQ between two structures in a.u.:110.4249
deltaQ between two structures in amu^1/2*Angs: 1.369
-----------------------------------------------
Generating structures...
transition level is 1.0765 eV
E_zpl ( Energy of zero phonon line ) is 1.0765 eV
Analysing deltaQ (the structure difference in generalized coordinate) ...
deltaQ between two structures in a.u.:110.4249
deltaQ between two structures in amu^1/2*Angs: 1.369
-----------------------------------------------
Generating structures...
-----------------------------------------------
..
-----------------------------------------------
calculation of initial state and final state all finished
-----------------------------------------------
analysing for nonradiative carrier capture coefficient...
electron-phonon coupling constant in eV/(Angs*amu^1/2): 0.1339
barrier for the nonradiative process is 0.70622 eV
try to plot the ccdiagram...
ccdiagram.png saved in dir /data2/home/…/cdc/C_N1/Nonrad_calc/_q-1_to_q0_
initial state phonon energy and wavefunction saved in dir /data2/home/…/cdc/C_N1/Nonrad_calc/_q-1_to_q0_/initial_state/phonon
final state phonon energy and wavefunction saved in dir /data2/home/…/cdc/C_N1/Nonrad_calc/_q-1_to_q0_/final_state/phonon
effective phonon energy of the final state is 0.04105 eV
Huang-Rhys factor of the final state is 8.92799
effective phonon energy of the initial state is 0.04671 eV
gaussian smearing is 0.03503 eV. The sommerfeld factor at 300.0 (K) is 11.50398 .
The carrier capture coefficient at 300.0 (K) is 8.94773e-11 cm^3/s.
see file nonradiative_rate.dat in dir /data2/home/…/cdc for more details.
coefficient.png saved in dir /data2/home/…/cdc/C_N1/Nonrad_calc/_q-1_to_q0_
--------------------- End of Calculation for Nonradiative Capture Rate ---------------------
dasp.in 文件如下:############## CDC Module ###############
cdc_job = nonrad_rate
cdc_defect = C_N1
cdc_charge = -1 0 #俘获前带电状态在前,俘获后带电状态在后
cdc_temperature = 300
cdc_band = 253 256 #带边的序号在前,缺陷能级的序号在后
spin_channel = 2
fitting_method = spline
dasp 5 执行CDC模块。等待期间无需额外操作。CDC模块会分析HSE泛函优化的初态和末态两个结构在广义坐标下的差异 \(ΔQ\) ,并沿着该方向线性地在更大的范围内产生一系列结构。
完成所有静态计算后,CDC模块可以根据产生的结构及对应的缺陷形成能大小得到初态与末态下的有效声子能量、声子波函数以及末态的黄-里斯因子(Huang-Rhys factor)以及用于估算非辐射俘获系数的 \(δ\) 函数的高斯展宽(gaussian smearing)和索莫菲因子(sommerfeld factor),还可以获得该缺陷初态与末态的一维位形图,输出为图片 ccdiagram.png ,如下所示。
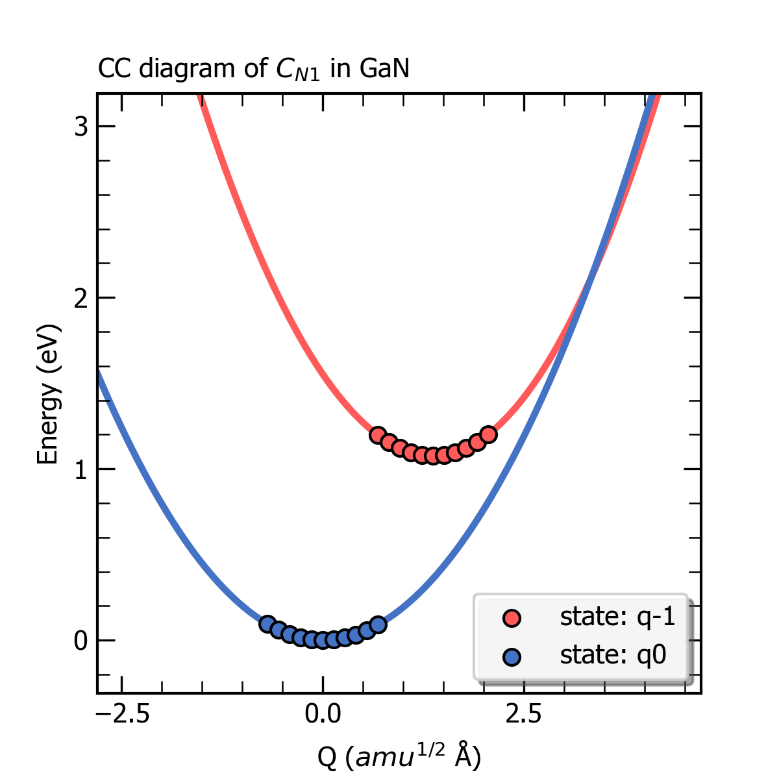
GaN中缺陷C_N1从-1价到0价的一维位形图。
可以看到,相比parabolic方法,采用spline的拟合方法将使得俘获势垒增加将近0.3 eV。我们建议总是优先采用parabolic方法进行势能面拟合。
最后,CDC模块会根据超胞体积、载流子有效质量等数据结合非辐射俘获系数的公式计算输出室温下该系数的大小,并在目录 /cdc 下输出 nonradiative_rate.dat 文件和图片 coefficient.png ,如下所示,其中给出了非辐射俘获系数以及非辐射俘获截面随温度的变化。
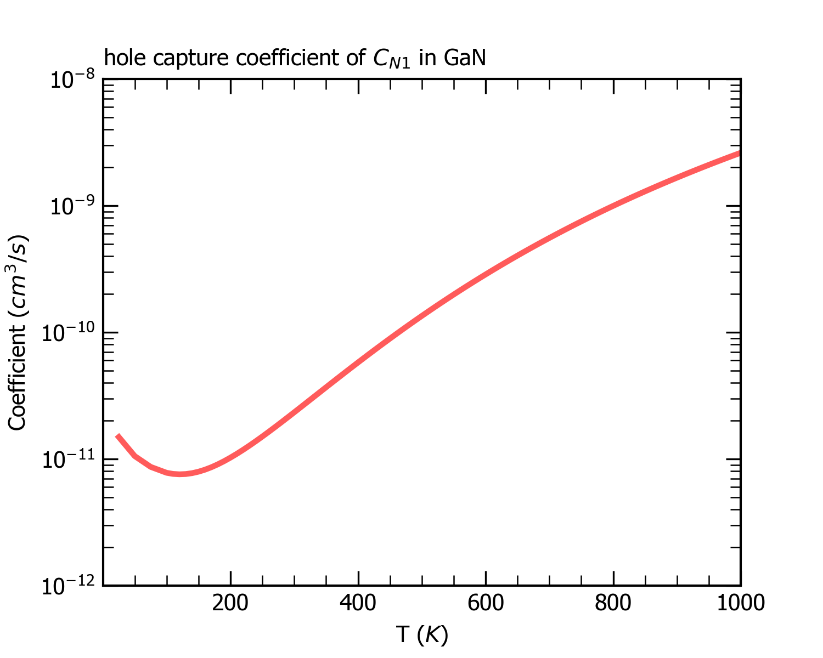
GaN中缺陷C_N1从-1价到0价空穴俘获系数随温度的变化关系。
5cdc.out :Analysing deltaQ (the structure difference in generalized coordinate) ...
deltaQ between two structures in a.u.:110.4249
deltaQ between two structures in amu^1/2*Angs: 1.369
-----------------------------------------------
Generating structures...
transition level is 1.0765 eV
E_zpl ( Energy of zero phonon line ) is 1.0765 eV
Analysing deltaQ (the structure difference in generalized coordinate) ...
deltaQ between two structures in a.u.:110.4249
deltaQ between two structures in amu^1/2*Angs: 1.369
-----------------------------------------------
Generating structures...
-----------------------------------------------
..
-----------------------------------------------
-----------------------------------------------
calculation of initial state and final state all finished
-----------------------------------------------
analysing for nonradiative carrier capture coefficient...
electron-phonon coupling constant in eV/(Angs*amu^1/2): 0.1339
barrier for the nonradiative process is 1.03875 eV
try to plot the ccdiagram...
ccdiagram.png saved in dir /data2/home/chensy/.../cdc/C_N1/Nonrad_calc/_q-1_to_q0_
initial state phonon energy and wavefunction saved in dir /data2/home/chensy/.../cdc/C_N1/Nonrad_calc/_q-1_to_q0_/initial_state/phonon
final state phonon energy and wavefunction saved in dir /data2/home/chensy/.../cdc/C_N1/Nonrad_calc/_q-1_to_q0_/final_state/phonon
effective phonon energy of the final state is 0.04108 eV
Huang-Rhys factor of the final state is 8.77443
effective phonon energy of the initial state is 0.04662 eV
gaussian smearing is 0.03497 eV. The sommerfeld factor at 300.0 (K) is 11.50398 .
The carrier capture coefficient at 300.0 (K) is 2.33508e-11 cm^3/s.
see file nonradiative_rate.dat in dir /data2/home/chensy/.../cdc for more details.
coefficient.png saved in dir /data2/home/chensy/.../cdc/C_N1/Nonrad_calc/_q-1_to_q0_
--------------------- End of Calculation for Nonradiative Capture Rate ---------------------
- 1
Audrius Alkauskas, Qimin Yan, and Chris G Van de Walle. First-principles theory of nonradiative carrier capture via multiphonon emission. Physical Review B, 90(7):075202, 2014. doi:10.1103/PhysRevB.90.075202.
5.7. ZnGeP2的复合缺陷计算¶
复合缺陷的计算需要在点缺陷计算完成之后进行,我们在5.4部份已经完成了ZnGeP2中点缺陷的计算,本部分是在此基础上完成复合缺陷的计算。
5.7.1. ZnGeP2复合缺陷计算设置¶
确定可能的复合缺陷名称
在完成ZnGeP2中点缺陷的计算之后,在 dec.out 文件中会输出可能的复合缺陷:
| 以下内容来自点缺陷计算DEC模块的程序日志 3dec.out :
Possible defect complexes:
1 Ge_Zn1+P_Ge1
2 Ge_P1+P_Ge1
3 Ge_P1+[Ge_Zn1+Zn_Ge1]-1
4 Ge_P1+Zn_Ge1
5 P_Ge1+[Ge_Zn1+Zn_Ge1]-1
6 V_Zn1+Zn_Ge1
7 P_Ge1+Zn_Ge1
8 Zn_Ge1+[Ge_Zn1+Zn_Ge1]-1
设置要计算的复合缺陷
如果你需要计算ZnGeP2中复合缺陷的性质,只需要在 dasp.in 中加入 complex 选项即可, 注意为了防止前后不一致,此处建议用户直接拷贝点缺陷计算完成后 3dec.out 文件中给出的复合缺陷名称。例如此处我们仅计算 \(Ge_{Zn1}+Zn_{Ge1}\) 和 \(Ge_{P1}+P_{Ge1}\) 两个复合缺陷,仅需在 dasp.in 中设置:
complex = Ge_Zn1+Zn_Ge1 Ge_P1+P_Ge1
计算复合缺陷
使用命令 dasp 3 继续执行DEC模块。
5.7.2. ZnGeP2中性复合缺陷结构优化及总能计算流程¶
产生复合缺陷结构
对于 \(Ge_{Zn1}+Zn_{Ge1}\) 复合缺陷,DASP会在 \(Ge_{Zn1}\) 缺陷附近寻找 最近邻, 次近邻 以及 次次近邻 的 \(Zn_{Ge1}\) 缺陷来形成复合缺陷,三种复合缺陷构型分别为 nn1, nn2 以及 nn3
############ Neutral Defect module start ############
Start generating neutral complex defect
Generate neutral defect at: Ge_Zn1+Zn_Ge1/nn1/initial_structure/q0
Generate neutral defect at: Ge_Zn1+Zn_Ge1/nn2/initial_structure/q0
Generate neutral defect at: Ge_Zn1+Zn_Ge1/nn3/initial_structure/q0
DASP产生的三种 \(Ge_{Zn1}+Zn_{Ge1}\) 复合缺陷构型。
同样,对于 \(Ge_{P1}+P_{Ge1}\) 复合缺陷,DASP同样会产生 nn1, nn2 以及 nn3 三种结构
Generate neutral defect at: Ge_P1+P_Ge1/nn1/initial_structure/q0
Generate neutral defect at: Ge_P1+P_Ge1/nn2/initial_structure/q0
Generate neutral defect at: Ge_P1+P_Ge1/nn3/initial_structure/q0
Neutral complex defect generation completed
DASP产生的三种 \(Ge_{P1}+P_{Ge1}\) 复合缺陷构型。
至此中性复合缺陷结构的产生完成。
检查中性点缺陷的结构优化是否完成
如果中性点缺陷计算完成,DASP则会自动跳过
Intrinsic defect directory Intrinsic_Defect exist, skip creating directory
Host directory in Intrinsic_Defect exist, skip creating directory
Start generating neutral vacancy defect
Neutral defect exist at: V_Zn1/initial_structure/q0, skipped
Neutral defect exist at: V_Ge1/initial_structure/q0, skipped
Neutral defect exist at: V_P1/initial_structure/q0, skipped
Neutral vacancy defect generation completed
Start generating neutral intrinsic antisite defect
Neutral defect exist at: Ge_Zn1/initial_structure/q0, skipped
Neutral defect exist at: P_Zn1/initial_structure/q0, skipped
Neutral defect exist at: Zn_Ge1/initial_structure/q0, skipped
Neutral defect exist at: P_Ge1/initial_structure/q0, skipped
Neutral defect exist at: Zn_P1/initial_structure/q0, skipped
Neutral defect exist at: Ge_P1/initial_structure/q0, skipped
Neutral intrinsic antisite defect generation completed
Start generating neutral intrinsic interstitial defect
Neutral defect exist at: Zn_i/random1/initial_structure/q0, skipped
Neutral defect exist at: Zn_i/random2/initial_structure/q0, skipped
Neutral defect exist at: Zn_i/random3/initial_structure/q0, skipped
Neutral defect exist at: Zn_i/random4/initial_structure/q0, skipped
Neutral defect exist at: Zn_i/random5/initial_structure/q0, skipped
Neutral defect exist at: Zn_i/random6/initial_structure/q0, skipped
Neutral defect exist at: Ge_i/random1/initial_structure/q0, skipped
Neutral defect exist at: Ge_i/random2/initial_structure/q0, skipped
Neutral defect exist at: Ge_i/random3/initial_structure/q0, skipped
Neutral defect exist at: Ge_i/random4/initial_structure/q0, skipped
Neutral defect exist at: Ge_i/random5/initial_structure/q0, skipped
Neutral defect exist at: Ge_i/random6/initial_structure/q0, skipped
Neutral defect exist at: P_i/random1/initial_structure/q0, skipped
Neutral defect exist at: P_i/random2/initial_structure/q0, skipped
Neutral defect exist at: P_i/random3/initial_structure/q0, skipped
Neutral defect exist at: P_i/random4/initial_structure/q0, skipped
Neutral defect exist at: P_i/random5/initial_structure/q0, skipped
Neutral defect exist at: P_i/random6/initial_structure/q0, skipped
Neutral intrinsic interstitial defect generation completed
############ Neutral Defect module end ############
提交任务优化中性复合缺陷的结构并完成总能的计算
结构产生完成后,DASP会自动提交任务优化中性复合缺陷的结构并完成总能的计算。
############ AutoRun module start ############
Job ****** submitted: /data/home/test/Zn_Ge_P/DASP-test-0128/dec/Intrinsic_Defect/Ge_Zn1+Zn_Ge1/nn2/initial_structure/q0
Job ****** submitted: /data/home/test/Zn_Ge_P/DASP-test-0128/dec/Intrinsic_Defect/Ge_Zn1+Zn_Ge1/nn1/initial_structure/q0
Job ****** submitted: /data/home/test/Zn_Ge_P/DASP-test-0128/dec/Intrinsic_Defect/Ge_Zn1+Zn_Ge1/nn3/initial_structure/q0
Job ****** submitted: /data/home/test/Zn_Ge_P/DASP-test-0128/dec/Intrinsic_Defect/Ge_P1+P_Ge1/nn2/initial_structure/q0
Job ****** submitted: /data/home/test/Zn_Ge_P/DASP-test-0128/dec/Intrinsic_Defect/Ge_P1+P_Ge1/nn1/initial_structure/q0
Job ****** submitted: /data/home/test/Zn_Ge_P/DASP-test-0128/dec/Intrinsic_Defect/Ge_P1+P_Ge1/nn3/initial_structure/q0
Job ****** submitted: /data/home/test/Zn_Ge_P/DASP-test-0128/dec/Intrinsic_Defect/Ge_Zn1+Zn_Ge1/nn2/initial_structure/q0/static
Job ****** submitted: /data/home/test/Zn_Ge_P/DASP-test-0128/dec/Intrinsic_Defect/Ge_Zn1+Zn_Ge1/nn1/initial_structure/q0/static
Job ****** submitted: /data/home/test/Zn_Ge_P/DASP-test-0128/dec/Intrinsic_Defect/Ge_Zn1+Zn_Ge1/nn3/initial_structure/q0/static
Job ****** submitted: /data/home/test/Zn_Ge_P/DASP-test-0128/dec/Intrinsic_Defect/Ge_P1+P_Ge1/nn2/initial_structure/q0/static
Job ****** submitted: /data/home/test/Zn_Ge_P/DASP-test-0128/dec/Intrinsic_Defect/Ge_P1+P_Ge1/nn1/initial_structure/q0/static
Job ****** submitted: /data/home/test/Zn_Ge_P/DASP-test-0128/dec/Intrinsic_Defect/Ge_P1+P_Ge1/nn3/initial_structure/q0/static
############ AutoRun module end ############
至此中性复合缺陷的结构优化及总能计算完成。
5.7.3. ZnGeP2带电复合缺陷结构优化及总能计算流程¶
检查ZnGeP2中带电缺陷的结构优化是否完成
如果带电点缺陷的结构优化及总能计算已经完成,DASP则会自动跳过
############ Ionized Defect module start ############
Ionized defect exist at: /data/home/test/Zn_Ge_P/DASP-test-0128/dec/Intrinsic_Defect/P_Zn1/initial_structure, skipped
Ionized defect exist at: /data/home/test/Zn_Ge_P/DASP-test-0128/dec/Intrinsic_Defect/Zn_P1/initial_structure, skipped
Ionized defect exist at: /data/home/test/Zn_Ge_P/DASP-test-0128/dec/Intrinsic_Defect/Ge_P1/initial_structure, skipped
...
ZnGeP2带电复合缺陷结构产生
DASP会根据中性复合缺陷的计算结果判断可能的带电状态并完成带电复合缺陷的结构优化及总能的计算:
Start generating ionized defects
Ionized defect path: /data/home/test/Zn_Ge_P/DASP-test-0128/dec/Intrinsic_Defect/Ge_Zn1+Zn_Ge1/nn2/initial_structure/q-4
Ionized defect path: /data/home/test/Zn_Ge_P/DASP-test-0128/dec/Intrinsic_Defect/Ge_Zn1+Zn_Ge1/nn2/initial_structure/q-3
Ionized defect path: /data/home/test/Zn_Ge_P/DASP-test-0128/dec/Intrinsic_Defect/Ge_Zn1+Zn_Ge1/nn2/initial_structure/q-2
Ionized defect path: /data/home/test/Zn_Ge_P/DASP-test-0128/dec/Intrinsic_Defect/Ge_Zn1+Zn_Ge1/nn2/initial_structure/q-1
Ionized defect path: /data/home/test/Zn_Ge_P/DASP-test-0128/dec/Intrinsic_Defect/Ge_Zn1+Zn_Ge1/nn2/initial_structure/q+1
Ionized defect path: /data/home/test/Zn_Ge_P/DASP-test-0128/dec/Intrinsic_Defect/Ge_Zn1+Zn_Ge1/nn2/initial_structure/q+2
Ionized defect path: /data/home/test/Zn_Ge_P/DASP-test-0128/dec/Intrinsic_Defect/Ge_Zn1+Zn_Ge1/nn2/initial_structure/q+3
Ionized defect path: /data/home/test/Zn_Ge_P/DASP-test-0128/dec/Intrinsic_Defect/Ge_Zn1+Zn_Ge1/nn2/initial_structure/q+4
Ionized defects generation completed
Start generating ionized defects
Ionized defect path: /data/home/test/Zn_Ge_P/DASP-test-0128/dec/Intrinsic_Defect/Ge_Zn1+Zn_Ge1/nn1/initial_structure/q-4
Ionized defect path: /data/home/test/Zn_Ge_P/DASP-test-0128/dec/Intrinsic_Defect/Ge_Zn1+Zn_Ge1/nn1/initial_structure/q-3
Ionized defect path: /data/home/test/Zn_Ge_P/DASP-test-0128/dec/Intrinsic_Defect/Ge_Zn1+Zn_Ge1/nn1/initial_structure/q-2
Ionized defect path: /data/home/test/Zn_Ge_P/DASP-test-0128/dec/Intrinsic_Defect/Ge_Zn1+Zn_Ge1/nn1/initial_structure/q-1
Ionized defect path: /data/home/test/Zn_Ge_P/DASP-test-0128/dec/Intrinsic_Defect/Ge_Zn1+Zn_Ge1/nn1/initial_structure/q+1
Ionized defect path: /data/home/test/Zn_Ge_P/DASP-test-0128/dec/Intrinsic_Defect/Ge_Zn1+Zn_Ge1/nn1/initial_structure/q+2
Ionized defect path: /data/home/test/Zn_Ge_P/DASP-test-0128/dec/Intrinsic_Defect/Ge_Zn1+Zn_Ge1/nn1/initial_structure/q+3
Ionized defect path: /data/home/test/Zn_Ge_P/DASP-test-0128/dec/Intrinsic_Defect/Ge_Zn1+Zn_Ge1/nn1/initial_structure/q+4
Ionized defects generation completed
Start generating ionized defects
Ionized defect path: /data/home/test/Zn_Ge_P/DASP-test-0128/dec/Intrinsic_Defect/Ge_Zn1+Zn_Ge1/nn3/initial_structure/q-4
Ionized defect path: /data/home/test/Zn_Ge_P/DASP-test-0128/dec/Intrinsic_Defect/Ge_Zn1+Zn_Ge1/nn3/initial_structure/q-3
Ionized defect path: /data/home/test/Zn_Ge_P/DASP-test-0128/dec/Intrinsic_Defect/Ge_Zn1+Zn_Ge1/nn3/initial_structure/q-2
Ionized defect path: /data/home/test/Zn_Ge_P/DASP-test-0128/dec/Intrinsic_Defect/Ge_Zn1+Zn_Ge1/nn3/initial_structure/q-1
Ionized defect path: /data/home/test/Zn_Ge_P/DASP-test-0128/dec/Intrinsic_Defect/Ge_Zn1+Zn_Ge1/nn3/initial_structure/q+1
Ionized defect path: /data/home/test/Zn_Ge_P/DASP-test-0128/dec/Intrinsic_Defect/Ge_Zn1+Zn_Ge1/nn3/initial_structure/q+2
Ionized defect path: /data/home/test/Zn_Ge_P/DASP-test-0128/dec/Intrinsic_Defect/Ge_Zn1+Zn_Ge1/nn3/initial_structure/q+3
Ionized defect path: /data/home/test/Zn_Ge_P/DASP-test-0128/dec/Intrinsic_Defect/Ge_Zn1+Zn_Ge1/nn3/initial_structure/q+4
Ionized defects generation completed
...
提交任务优化带电复合缺陷的结构并完成总能的计算
############ AutoRun module start ############
Job ****** submitted: /data/home/test/Zn_Ge_P/DASP-test-0128/dec/Intrinsic_Defect/Ge_Zn1+Zn_Ge1/nn2/initial_structure/q-1
Job ****** submitted: /data/home/test/Zn_Ge_P/DASP-test-0128/dec/Intrinsic_Defect/Ge_Zn1+Zn_Ge1/nn2/initial_structure/q-4
Job ****** submitted: /data/home/test/Zn_Ge_P/DASP-test-0128/dec/Intrinsic_Defect/Ge_Zn1+Zn_Ge1/nn2/initial_structure/q-2
Job ****** submitted: /data/home/test/Zn_Ge_P/DASP-test-0128/dec/Intrinsic_Defect/Ge_Zn1+Zn_Ge1/nn2/initial_structure/q+3
Job ****** submitted: /data/home/test/Zn_Ge_P/DASP-test-0128/dec/Intrinsic_Defect/Ge_Zn1+Zn_Ge1/nn2/initial_structure/q-3
Job ****** submitted: /data/home/test/Zn_Ge_P/DASP-test-0128/dec/Intrinsic_Defect/Ge_Zn1+Zn_Ge1/nn2/initial_structure/q+1
Job ****** submitted: /data/home/test/Zn_Ge_P/DASP-test-0128/dec/Intrinsic_Defect/Ge_Zn1+Zn_Ge1/nn2/initial_structure/q+4
Job ****** submitted: /data/home/test/Zn_Ge_P/DASP-test-0128/dec/Intrinsic_Defect/Ge_Zn1+Zn_Ge1/nn2/initial_structure/q+2
Job ****** submitted: /data/home/test/Zn_Ge_P/DASP-test-0128/dec/Intrinsic_Defect/Ge_Zn1+Zn_Ge1/nn1/initial_structure/q-1
Job ****** submitted: /data/home/test/Zn_Ge_P/DASP-test-0128/dec/Intrinsic_Defect/Ge_Zn1+Zn_Ge1/nn1/initial_structure/q-4
Job ****** submitted: /data/home/test/Zn_Ge_P/DASP-test-0128/dec/Intrinsic_Defect/Ge_Zn1+Zn_Ge1/nn1/initial_structure/q-2
Job ****** submitted: /data/home/test/Zn_Ge_P/DASP-test-0128/dec/Intrinsic_Defect/Ge_Zn1+Zn_Ge1/nn1/initial_structure/q+3
Job ****** submitted: /data/home/test/Zn_Ge_P/DASP-test-0128/dec/Intrinsic_Defect/Ge_Zn1+Zn_Ge1/nn1/initial_structure/q-3
Job ****** submitted: /data/home/test/Zn_Ge_P/DASP-test-0128/dec/Intrinsic_Defect/Ge_Zn1+Zn_Ge1/nn1/initial_structure/q+1
...
5.7.4. ZnGeP2复合缺陷形成能计算及绘图¶
ZnGeP2复合缺陷形成能计算
优化完各个价态结构并计算得到总能后,DASP会计算所有缺陷各个价态的形成能及转变能级。
############ Formation Energy module start ############
Start the formation energy calculation
The formation energy (neutral) of P_Zn1 at p1 is 3.993 eV
The formation energy (neutral) of P_Zn1 at p2 is 2.591 eV
The formation energy (neutral) of P_Zn1 at p3 is 2.751 eV
The formation energy (neutral) of P_Zn1 at p4 is 3.794 eV
The potential alignment (q = -1) q_deltaV = 0.014 eV, the image charge correction E_correct = 0.07 eV
The transition level (-/0) above VBM: 1.373 eV
The potential alignment (q = -4) q_deltaV = 0.4 eV, the image charge correction E_correct = 1.117 eV
The transition level (4-/0) above VBM: 2.057 eV
The potential alignment (q = -2) q_deltaV = 0.041 eV, the image charge correction E_correct = 0.279 eV
The transition level (2-/0) above VBM: 1.83 eV
The potential alignment (q = 3) q_deltaV = 0.555 eV, the image charge correction E_correct = 0.628 eV
The transition level (0/3+) above VBM: 0.573 eV
The potential alignment (q = -3) q_deltaV = 0.366 eV, the image charge correction E_correct = 0.628 eV
The transition level (3-/0) above VBM: 1.87 eV
The potential alignment (q = 1) q_deltaV = 0.09 eV, the image charge correction E_correct = 0.07 eV
The transition level (0/+) above VBM: 1.075 eV
The potential alignment (q = 4) q_deltaV = 0.542 eV, the image charge correction E_correct = 1.117 eV
The transition level (0/4+) above VBM: 0.319 eV
The potential alignment (q = 2) q_deltaV = 0.28 eV, the image charge correction E_correct = 0.279 eV
The transition level (0/2+) above VBM: 0.76 eV
The formation energy calculation completed
...
############ Formation Energy module end ############
ZnGeP2复合缺陷形成能绘图
完成形成能的计算后,DASP会绘制所有缺陷(包括点缺陷,复合缺陷)在特定化学势条件下的形成能随费米能级变化图。
############ Plot Diagram module start ############
Formation energy calculation not done: /data/home/test/Zn_Ge_P/DASP-test-0128/dec/Intrinsic_Defect/P_i/random6/initial_structure, skipped
Formation energy calculation not done: /data/home/test/Zn_Ge_P/DASP-test-0128/dec/Intrinsic_Defect/P_i/random5/initial_structure, skipped
Formation energy calculation not done: /data/home/test/Zn_Ge_P/DASP-test-0128/dec/Intrinsic_Defect/P_i/random2/initial_structure, skipped
Formation energy calculation not done: /data/home/test/Zn_Ge_P/DASP-test-0128/dec/Intrinsic_Defect/Ge_i/random6/initial_structure, skipped
Formation energy calculation not done: /data/home/test/Zn_Ge_P/DASP-test-0128/dec/Intrinsic_Defect/Ge_i/random4/initial_structure, skipped
Formation energy calculation not done: /data/home/test/Zn_Ge_P/DASP-test-0128/dec/Intrinsic_Defect/Ge_i/random2/initial_structure, skipped
Formation energy calculation not done: /data/home/test/Zn_Ge_P/DASP-test-0128/dec/Intrinsic_Defect/Zn_i/random3/initial_structure, skipped
Formation energy calculation not done: /data/home/test/Zn_Ge_P/DASP-test-0128/dec/Intrinsic_Defect/Zn_i/random1/initial_structure, skipped
Formation energy calculation not done: /data/home/test/Zn_Ge_P/DASP-test-0128/dec/Intrinsic_Defect/Zn_i/random2/initial_structure, skipped
Start plotting the diagrams
Generate formation energy diagrams at p1: /data/home/test/Zn_Ge_P/DASP-test-0128/dec/Formation_Energy_Intrinsic_Defect/p1.dat
Generate formation energy diagrams at p2: /data/home/test/Zn_Ge_P/DASP-test-0128/dec/Formation_Energy_Intrinsic_Defect/p2.dat
Generate formation energy diagrams at p3: /data/home/test/Zn_Ge_P/DASP-test-0128/dec/Formation_Energy_Intrinsic_Defect/p3.dat
Generate formation energy diagrams at p4: /data/home/test/Zn_Ge_P/DASP-test-0128/dec/Formation_Energy_Intrinsic_Defect/p4.dat
Generate transition level diagram: /data/home/test/Zn_Ge_P/DASP-test-0128/dec/Transition_Level_Intrinsic_Defect/tl.dat
All diagrams completed
Possible defect complexes:
1 Ge_Zn1+P_Ge1
2 Ge_P1+P_Ge1
3 Ge_P1+[Ge_Zn1+Zn_Ge1]-1
4 Ge_P1+Zn_Ge1
5 P_Ge1+[Ge_Zn1+Zn_Ge1]-1
6 V_Zn1+Zn_Ge1
7 P_Ge1+Zn_Ge1
8 Zn_Ge1+[Ge_Zn1+Zn_Ge1]-1
############ Plot Diagram module end ############
DASP会绘制所有缺陷的形成能随费米能级的变化图,此处仅显示了所考虑的复合缺陷及与之相关的点缺陷形成能随费米能级变化图。
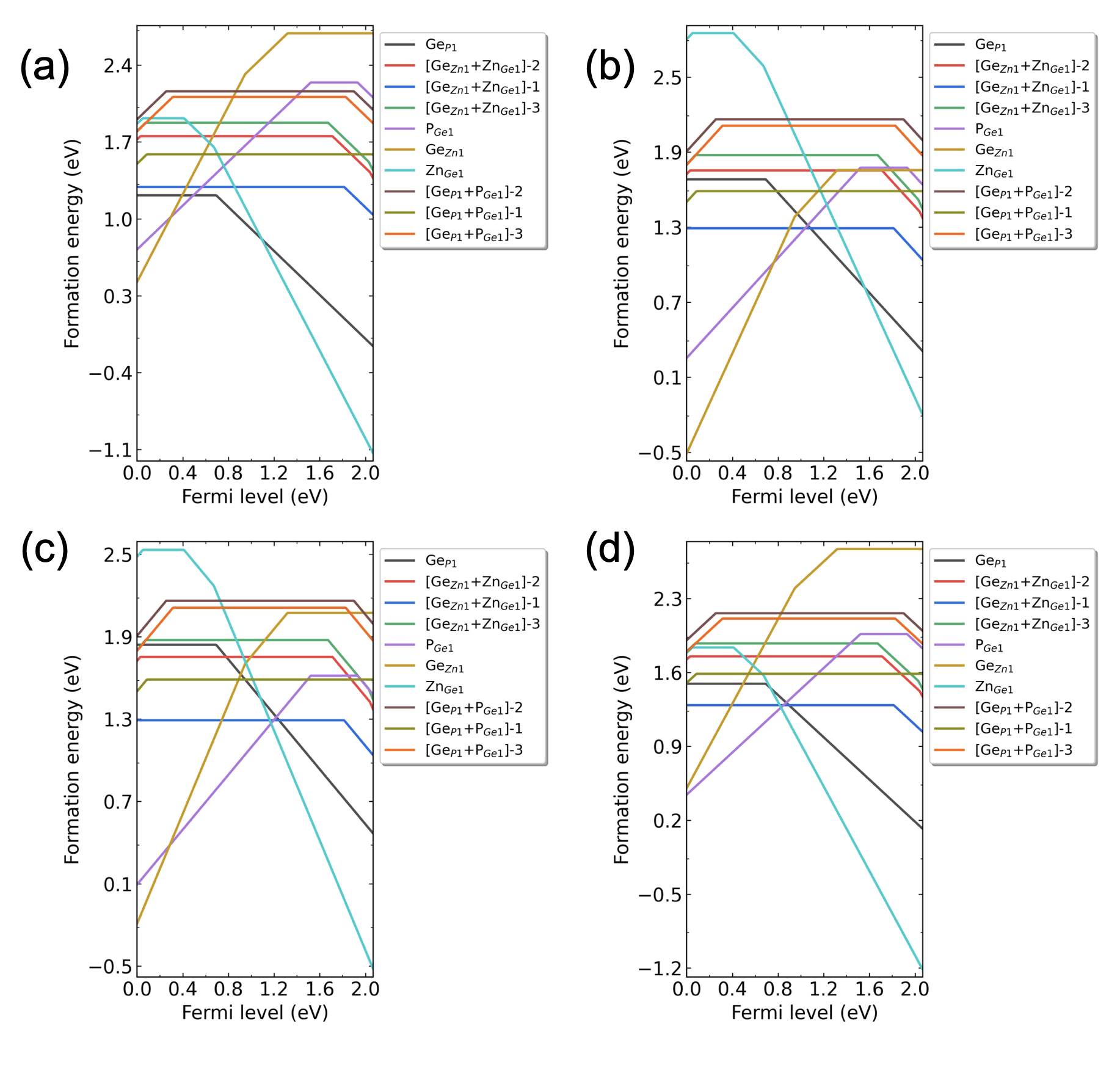
ZnGeP2中复合缺陷及对应点缺陷在化学势点(a) p1, (b) p2, (c) p3, (d) p4 处的形成能随费米能级的变化。
从图中可以发现:对于这两类复合缺陷,总是最近邻构型(即 nn1 )的形成能更低。
至此DASP已经完成了用户设置的所有缺陷的形成能、转变能级的计算,如果要完成缺陷(杂质)浓度及载流子动力学的计算,继续执行 dasp 4 以及 dasp 5 即可。
5.8. GaN中本征点缺陷和复合缺陷计算¶
5.8.1. 准备计算PREPARE¶
5.8.1.1. 准备POSCAR与dasp.in¶
Materials Project 数据库中找到GaN原胞的POSCAR结构,显示如下: Ga2 N2
1.0
3.2162899971 0.0000000000 0.0000000000
-1.6081449986 2.7853888434 0.0000000000
0.0000000000 0.0000000000 5.2399621010
Ga N
2 2
Direct
0.333333343 0.666666687 0.999119997
0.666666687 0.333333343 0.499119997
0.333333343 0.666666687 0.375880003
0.666666687 0.333333343 0.875880003
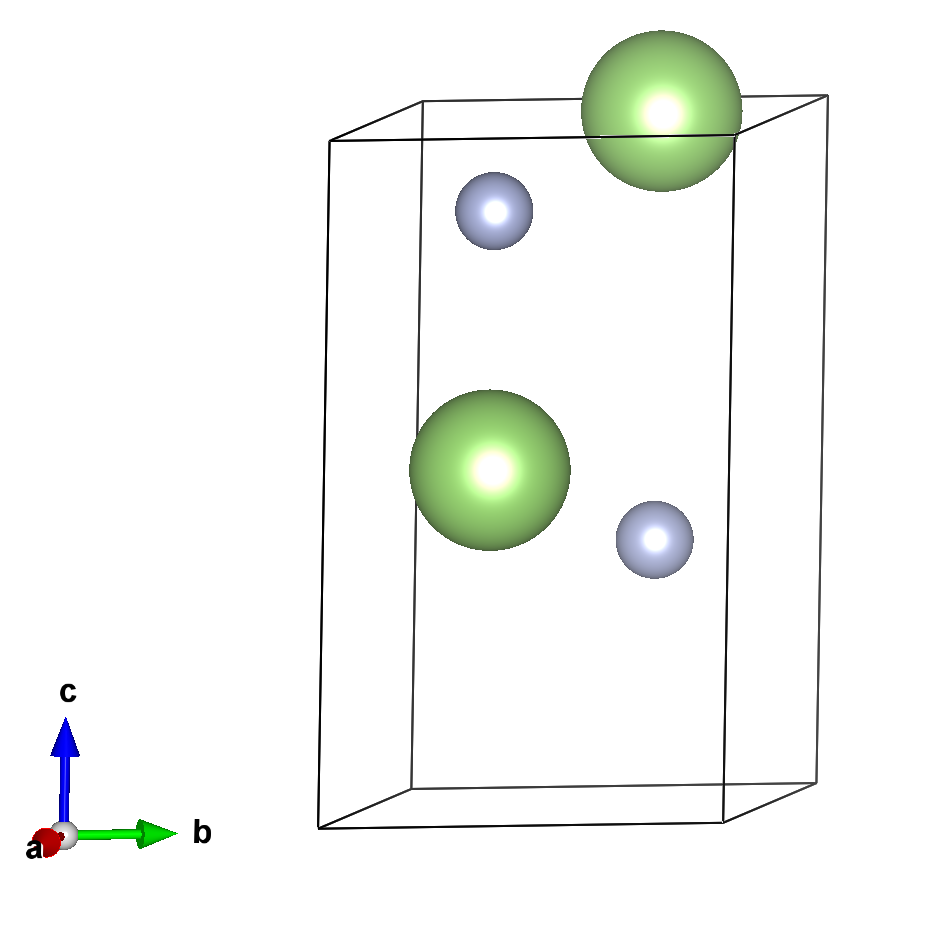
GaN的原胞结构。
dasp.in 中写入必要参数############## Job Scheduling ##############
cluster = SLURM # (job scheduling system)
node_number = 2 # (number of node)
core_per_node = 52 # (core per node)
queue = batch # (name of queue/partition)
max_time = 24:00:00 # (maximum time for a single DFT calculation)
vasp_path_dec = /opt/vasp.5.4.4/bin/vasp_gam # (path of VASP)
vasp_path_tsc = /opt/vasp.5.4.4/bin/vasp_std
job_name = submit_job # (name of script)
potcar_path = /opt/POT/potpaw_PBE # (path of pseudopotentials)
max_job = 5
############## TSC Module ##############
database_api = ******************* # (str-list type)
############## DEC Module ##############
level = 2 # (level=1: PBE+PBE; level=2: PBE+HSE; level=3: HSE+HSE)
min_atom = 128
max_atom = 128
intrinsic = T # (default: T)
vacancy= T # (default: T)
antisite = T # (default: T)
interstitial = F # (default: T)
correction = FNV # (default: none)
epsilon = 10.4
Eg_real = 3.5 # (experimental band gap)
dasp.in 中所有列出的参数进行说明。cluster = SLURM
# 表示使用集群的队列系统为SLURM
node_number = 2
# 对于每个单独的计算,使用2个节点
core_per_node = 52
# 对于每个节点,使用52个核。因此对于每个单独的计算,总共使用2*52=104核
queue = batch
# 使用名为"batch"的队列进行计算。因此,在设置dasp.in参数之前,需要确认超算/集群上的队列名、节点、核数
max_time = 24:00:00 # (maximum time for a single DFT calculation)
# 每个单独的计算所允许的最大时间为24小时,可任意设置。
vasp_path_dec = /opt/vasp.5.4.4/bin/vasp_gam # (path of VASP)
vasp_path_tsc = /opt/vasp.5.4.4/bin/vasp_std
# 对于TSC的计算,采用std版的VASP。对于DEC的单k点计算,采用gam版的VASP。
job_name = submit_job # (name of script)
# 提交任务的脚本,命名为"submit_job",可任意设置。
potcar_path = /opt/POT/potpaw_PBE # (path of pseudopotentials)
# POTCAR的路径
max_job = 5
# 允许同时在跑的任务最大数
database_api = ******************* # (str-list type)
# 用于访问Materials Project数据库
level = 2 # (level=1: PBE+PBE; level=2: PBE+HSE; level=3: HSE+HSE)
# 对于超胞,使用PBE优化其原子位置,使用HSE计算其总能
min_atom = 128
max_atom = 128
# 我们希望生成的超胞大小在128-128个原子之间,且尽量使a=b=c,a⊥b⊥c
intrinsic = T # (default: T)
# 产生本征缺陷,V_Ga V_N Ga_N N_Ga Ga_i N_i
vacancy = T # (default: T)
# 产生空位缺陷,V_Ga V_N
antisite = T # (default: T)
# 产生替位缺陷,Ga_N N_Ga
interstitial = F # (default: T)
# 不产生间隙缺陷
correction = FNV # (default: none)
# 带电缺陷的修正方案采用FNV修正
epsilon = 10.4
# CdTe的介电常数为10.4
Eg_real = 3.5 # (experimental band gap)
# GaN的实验带隙值约为3.5 eV,DASP将根据此数据调整AEXX参数,从而使得无缺陷超胞的带隙值等于3.5 eV
5.8.1.2. 使用DASP产生必要输入文件¶
dasp.in 文件,执行 dasp 1 ,即可启动PREPARE模块,此后无需额外操作。DASP会输出 1prepare.out 文件记录程序的运行日志。5.8.1.3. PREPARE模块运行流程¶
产生超胞:
POSCAR_nearlycube :Cubic_cell
1.0
11.0572163121 0.0000000000 0.0000000000
0.0000000000 12.1767486142 0.0000000000
0.0000000000 -0.6053683572 10.8724354477
Ga N
64 64
Direct
0.0833333333 0.1084788737 0.9056525825
0.0000000000 0.2334788737 0.1556525825
0.0833333333 0.4834788737 0.1556525825
0.0000000000 0.6084788737 0.4056525825
...
通过可视化软件,我们可以看到:给出的GaN原胞晶轴的夹角是较小的,但是经过DASP生成的超胞是三边垂直的。
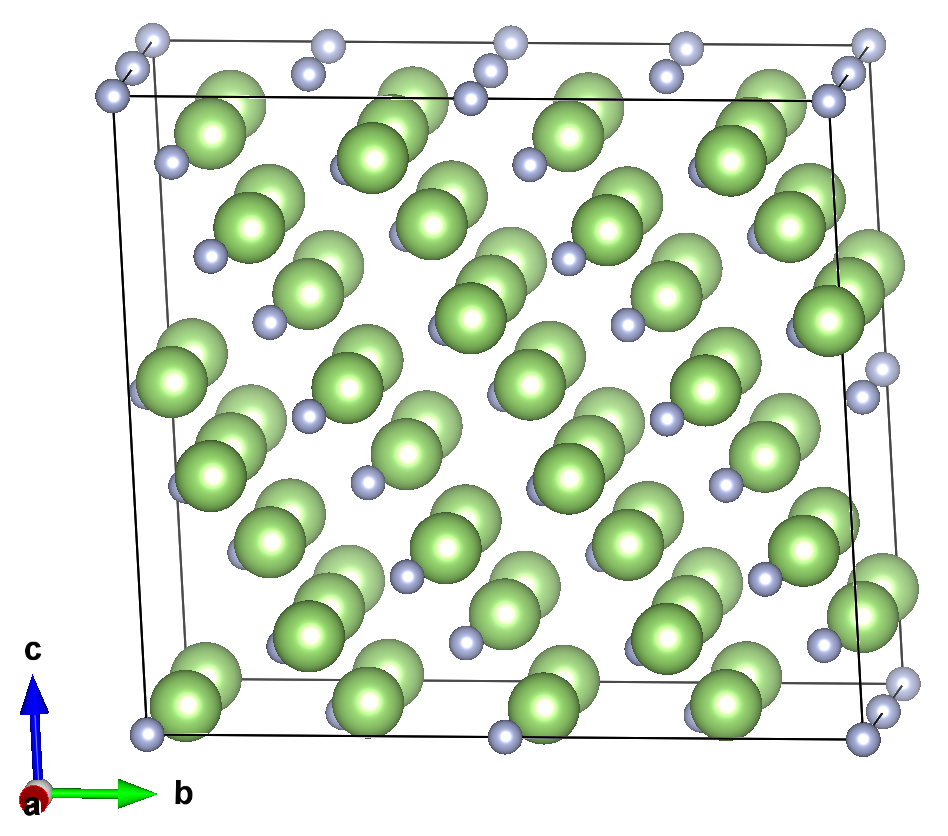
GaN的超胞结构。
马德隆常数计算:
1prepare.out 的输出如下:############ Prepare Files module start ############
Read the structure file POSCAR you provided
Get the primitive cell POSCAR_primitive from POSCAR
Generate the intermediate cell POSCAR_intermediate from POSCAR
Generate the nearlycube cell POSCAR_nearlycube
Generate job script through dasp.in parameters
Generate single-point KPOINTS
Generate pseudopotential file POTCAR through potcar_dir you set
Generate commonly used vasp input file INCAR
Start the madelung constant calculation
Generate the madelung calculation directory
Generate madelung calculation POSCAR
Generate madelung calculation POTCAR
Generate madelung calculation INCAR
Generate madelung calculation KPOINTS
Generate madelung calculation job script
Job 118920 submitted: /home/test/GaN/dec/madelung/static
The madelung constant calculation completed
The madelung constant = 2.828
HSE交换参数计算:
cd ./dec/AEXX
ls
0.25 0.3 AEXX.list
1prepare.out 可以看到如下日志:Start the HSE parameter AEXX calculation
Job 118924 submitted: /home/test/GaN/dec/AEXX/0.25/static
Job 118927 submitted: /home/test/GaN/dec/AEXX/0.3/static
The HSE parameter AEXX calculation completed
The HSE parameter AEXX = 0.3
level = 2: Generate PBE relax vasp input file INCAR-relax
level = 2: Generate HSE static vasp input file INCAR-static
host超胞原子位置的优化:
POSCAR_final 。同时也可以在 1prepare.out 可以DASP运行结束的标志,并告诉我们下一步需要做TSC模块的计算。Start the POSCAR_nearlycube relax calculation
Generate the POSCAR_nearlycube relax directory
Job 118940 submitted: /home/test/GaN/dec/relax
The POSCAR_nearlycube relax calculation completed
Get the final structure POSCAR_final
############ Prepare Files module end ############
PREPARE模块 finished, please run DASP-TSC next
5.8.2. 热力学稳定性和元素化学势计算TSC¶
5.8.2.1. 运行TSC模块¶
dasp 1 执行PREPARE模块时,会生成GaN/dec目录,并在该目录中产生 1prepare.out 文件。等待程序执行完毕, 1prepare.out 有相应的完成标志。进入GaN/dec目录。确认INCAR-relax,INCAR-static文件中的参数是可行的。(用户可修改INCAR,DASP将根据此目录中的INCAR做后续的计算)dasp 2 执行TSC模块。同样地,TSC模块会在GaN目录中生成名为tsc的目录,里面记录了TSC程序的计算输出,包括各计算目录以及运行日志文件 2tsc.out 。等待程序完成期间无需额外操作。5.8.2.2. TSC模块运行流程¶
host结构的总能计算(与MP参数保持一致):
Materials Project 数据库完全一致的输入参数(INCAR,KPOINTS,POTCAR)来对用户给定的原胞做结构优化和静态计算。因此,该计算得到的总能与MP数据库的总能是可比的。此步骤是为了得到影响GaN稳定性的 关键杂相 。通过目录可以看到:cd tsc
cd GaN/
ls
relaxation1 relaxation2 static
关键杂相判断:
2tsc.out 中可看到相关的信息:...
analysing the thermodynamic stability of GaN.
The stability of GaN is: True.
key phases of GaN are: Ga N2 .
file key_phases_info_recalc.yaml updated.
analysing of GaN is done.
...
host与杂相结构的总能计算(PREPARE模块确定的参数):
2tsc.out 如下:...
Job 118941 submitted: /home/test/GaN/tsc/GaN/static_recalc
Job 118942 submitted: /home/test/GaN/tsc/Ga/static_recalc
Job 118943 submitted: /home/test/GaN/tsc/N2/static_recalc
...
化学势的计算:
dasp.in :# 顺序与POSCAR中元素顺序一致,即第一列是Ga,第二列是N
E_pure = -4.0248 -12.0634
p1 = 0.0 -1.305
p2 = -1.305 0.0
2tsc.out 可以看到程序执行完毕的输出: dir '2d-figures','3d-figures','ori_data_MP' ready. analysing the thermodynamic stability of GaN.
The stability of GaN is: True.
key phases of GaN are: Ga N2 .
analysing of GaN is done.
sub-module of tsc: 'auto thermodynamic calculation' ends successfully.
DASP-TSC finished
5.8.3. 缺陷形成能和转变能级计算DEC¶
5.8.3.1. 运行DEC模块¶
dasp 2 执行TSC模块时,会生成GaN/tsc目录,并在该目录中产生 2tsc.out 文件。等待程序执行完毕, 2tsc.out 有相应的完成标志。打开GaN/dasp.in,确认化学势已被程序自动输入。dasp 3 执行DEC模块。DEC会在第一步已经生成的dec目录中继续输出相关文件,包括缺陷结构,缺陷目录,以及运行日志文件 3dec.out 。等待程序完成期间无需额外操作。5.8.3.2. DEC模块运行流程¶
产生缺陷结构:
dasp.in 中的参数,DEC模块将产生GaN的本征缺陷,即生成GaN/dec/Intrinsic_Defect计算目录,在其下面分别有空位缺陷V_Ga,V_N,替位缺陷Ga_N,N_Ga的缺陷结构和目录。根据对称性判aN断,GaN晶格中不存在非等价的Ga和N原子,因此同样的缺陷构型只需产生一种。cd dec/Intrinsic_Defect/
ls
Ga_N1 host Intrinsic_Defect.list N_Ga1 V_Ga1 V_N1
3dec.out 看到DEC模块的输出如下:############ Neutral Defect module start ############
Make intrinsic defect directory Intrinsic_Defect
Generate host directory in Intrinsic_Defect
Start generating neutral vacancy defect
Generate neutral defect at: V_Ga1/initial_structure/q0
Generate neutral defect at: V_N1/initial_structure/q0
Neutral vacancy defect generation completed
Start generating neutral intrinsic antisite defect
Generate neutral defect at: N_Ga1/initial_structure/q0
Generate neutral defect at: Ga_N1/initial_structure/q0
Neutral intrinsic antisite defect generation completed
############ Neutral Defect module end ############
提交各缺陷q=0计算任务:
dasp.in 中 level = 2 的参数),此步骤等待时间较长。可随时检查 3dec.out 文件。 3dec.out 中的相关信息如下所示:Job 119084 submitted: /home/test/GaN/dec/Intrinsic_Defect/V_N1/initial_structure/q0
Job 119086 submitted: /home/test/GaN/dec/Intrinsic_Defect/N_Ga1/initial_structure/q0
Job 119088 submitted: /home/test/GaN/dec/Intrinsic_Defect/V_Ga1/initial_structure/q0
Job 119090 submitted: /home/test/GaN/dec/Intrinsic_Defect/Ga_N1/initial_structure/q0
Job 119095 submitted: /home/test/GaN/dec/Intrinsic_Defect/host
Job 119097 submitted: /home/test/GaN/dec/Intrinsic_Defect/V_N1/initial_structure/q0/static
Job 119099 submitted: /home/test/GaN/dec/Intrinsic_Defect/N_Ga1/initial_structure/q0/static
Job 119101 submitted: /home/test/GaN/dec/Intrinsic_Defect/V_Ga1/initial_structure/q0/static
Job 119103 submitted: /home/test/GaN/dec/Intrinsic_Defect/Ga_N1/initial_structure/q0/static
产生带电缺陷的计算目录:
3dec.out 中的相关信息如下所示:############ Ionized Defect module start ############
Start generating ionized defects
Ionized defect path: /home/test/GaN/dec/Intrinsic_Defect/V_N1/initial_structure/q+1
Ionized defects generation completed
Start generating ionized defects
Ionized defect path: /home/test/GaN/dec/Intrinsic_Defect/N_Ga1/initial_structure/q-4
Ionized defect path: /home/test/GaN/dec/Intrinsic_Defect/N_Ga1/initial_structure/q-3
Ionized defect path: /home/test/GaN/dec/Intrinsic_Defect/N_Ga1/initial_structure/q-2
Ionized defect path: /home/test/GaN/dec/Intrinsic_Defect/N_Ga1/initial_structure/q-1
Ionized defect path: /home/test/GaN/dec/Intrinsic_Defect/N_Ga1/initial_structure/q+1
Ionized defect path: /home/test/GaN/dec/Intrinsic_Defect/N_Ga1/initial_structure/q+2
Ionized defect path: /home/test/GaN/dec/Intrinsic_Defect/N_Ga1/initial_structure/q+3
Ionized defect path: /home/test/GaN/dec/Intrinsic_Defect/N_Ga1/initial_structure/q+4
Ionized defects generation completed
...
提交各缺陷q≠0的计算任务:
dasp.in 中 level = 2 的参数),此步骤的等待时间比 q=0 计算时间更长。 3dec.out 中的相关信息如下所示:############ AutoRun - Ionized Defect module start ############
Job 119122 submitted: /home/test/GaN/dec/Intrinsic_Defect/N_Ga1/initial_structure/q-3
Job 119124 submitted: /home/test/GaN/dec/Intrinsic_Defect/N_Ga1/initial_structure/q+2
Job 119126 submitted: /home/test/GaN/dec/Intrinsic_Defect/N_Ga1/initial_structure/q-2
Job 119128 submitted: /home/test/GaN/dec/Intrinsic_Defect/N_Ga1/initial_structure/q-4
Job 119130 submitted: /home/test/GaN/dec/Intrinsic_Defect/N_Ga1/initial_structure/q+1
Job 119132 submitted: /home/test/GaN/dec/Intrinsic_Defect/N_Ga1/initial_structure/q+4
Job 119134 submitted: /home/test/GaN/dec/Intrinsic_Defect/N_Ga1/initial_structure/q+3
Job 119136 submitted: /home/test/GaN/dec/Intrinsic_Defect/N_Ga1/initial_structure/q-1
...
计算带电缺陷的修正:
dasp.in 中 correction = FNV 的参数),并计算其缺陷形成能和转变能级。每个缺陷各价态的修正量和形成能的具体数值,都记录在 3dec.out 中:...
The formation energy (neutral) of N_Ga1 at p1 is 10.177 eV
The formation energy (neutral) of N_Ga1 at p2 is 7.567 eV
The FNV correction (q = -3) E_correct = 1.307 eV
The transition level (3-/0) above VBM: 3.395 eV
The FNV correction (q = 2) E_correct = 0.629 eV
The transition level (0/2+) above VBM: 1.579 eV
The FNV correction (q = -2) E_correct = 0.899 eV
The transition level (2-/0) above VBM: 3.175 eV
The FNV correction (q = -4) E_correct = 2.202 eV
The transition level (4-/0) above VBM: 3.32 eV
The FNV correction (q = 1) E_correct = 0.113 eV
The transition level (0/+) above VBM: 1.494 eV
The FNV correction (q = 4) E_correct = 1.766 eV
The transition level (0/4+) above VBM: 0.459 eV
The FNV correction (q = 3) E_correct = 1.196 eV
The transition level (0/3+) above VBM: 0.767 eV
The FNV correction (q = -1) E_correct = 0.32 eV
The transition level (-/0) above VBM: 2.873 eV
...
defect.log 文件中。输出形成能图像:
GaN在(a)富Ga和(b)富N时的缺陷形成能随费米能级的变化。
至此GaN中的本征点缺陷计算已经完成,下面将继续进行GaN中复合缺陷的计算。值得一提的是,复合缺陷的计算必须在点缺陷计算完成之后进行。
5.8.4. GaN中复合缺陷计算¶
5.8.4.1. 复合缺陷计算设置¶
确定可能的复合缺陷名称
在GaN中点缺陷的计算完成后,在 3dec.out 文件中会输出可能的复合缺陷:
| 以下内容来自点缺陷计算DEC模块的程序日志 3dec.out :
Possible defect complexes:
1 V_N1+V_Ga1
2 V_N1+N_Ga1
3 Ga_N1+N_Ga1
设置要计算的复合缺陷
要计算GaN中复合缺陷的性质只需要在 dasp.in 中加入 complex 选项即可。 需要注意的是,复合缺陷的名字要与前面计算的点缺陷的名字保持一致。例如此处我们需要计算 \(V_{N1}+V_{N1}\) ,\(V_{Ga1}+V_{N1}\) ,\(Ga_{N1}+N_{Ga1}\) 四种复合缺陷,仅需在 dasp.in 中设置:
complex= V_N1+V_N1 V_Ga1+V_N1 Ga_N1+N_Ga1 V_N1+Ga_N1
计算复合缺陷
使用命令 dasp 3 继续执行DEC模块。
5.8.4.2. GaN中电中性复合缺陷结构优化及总能计算流程¶
产生复合缺陷结构
对于 \(V_{N1}+V_{N1}\) 复合缺陷,DASP会在 \(V_{N1}\) 缺陷附近寻找 最近邻, 次近邻 以及 次次近邻 的 \(V_{N1}\) 缺陷来形成复合缺陷,三种复合缺陷构型分别为 nn1, nn2 以及 nn3
############ Neutral Defect module start ############
Start generating neutral complex defect
Generate neutral defect at: V_N1+V_N1/nn1/initial_structure/q0
Generate neutral defect at: V_N1+V_N1/nn2/initial_structure/q0
Generate neutral defect at: V_N1+V_N1/nn3/initial_structure/q0
DASP产生的三种 \(V_{N1}+V_{N1}\) 复合缺陷构型。
同样,对另外三种复合缺陷,DASP同样会产生 nn1, nn2 以及 nn3 三种结构
Generate neutral defect at: V_Ga1+V_N1/nn1/initial_structure/q0
Generate neutral defect at: V_Ga1+V_N1/nn2/initial_structure/q0
Generate neutral defect at: V_Ga1+V_N1/nn3/initial_structure/q0
Generate neutral defect at: Ga_N1+N_Ga1/nn1/initial_structure/q0
Generate neutral defect at: Ga_N1+N_Ga1/nn2/initial_structure/q0
Generate neutral defect at: Ga_N1+N_Ga1/nn3/initial_structure/q0
Generate neutral defect at: Ga_N1+V_N1/nn1/initial_structure/q0
Generate neutral defect at: Ga_N1+V_N1/nn2/initial_structure/q0
Generate neutral defect at: Ga_N1+V_N1/nn3/initial_structure/q0
Neutral complex defect generation completed
至此电中性复合缺陷结构的产生完成。
检查电中性点缺陷的结构优化是否完成
在电中性复合缺陷计算时,程序也会检查电中性点缺陷的计算。若之前电中性点缺陷的计算已经完成,DASP则会自动跳过
Neutral complex defect generation completed
Intrinsic defect directory Intrinsic_Defect exist, skip creating directory
Host directory in Intrinsic_Defect exist, skip creating directory
Start generating neutral vacancy defect
Neutral defect exist at: V_Ga1/initial_structure/q0, skipped
Neutral defect exist at: V_N1/initial_structure/q0, skipped
Neutral vacancy defect generation completed
Start generating neutral intrinsic antisite defect
Neutral defect exist at: N_Ga1/initial_structure/q0, skipped
Neutral defect exist at: Ga_N1/initial_structure/q0, skipped
Neutral intrinsic antisite defect generation completed
############ Neutral Defect module end ############
提交任务优化中性复合缺陷的结构并完成总能的计算
结构产生完成后,DASP会自动提交任务优化中性复合缺陷的结构并完成总能的计算。
############ AutoRun module start ############
Job 119424 submitted: /data/home/test/GaN/dec/Intrinsic_Defect/V_N1+V_N1/nn2/initial_structure/q0
Job 119426 submitted: /data/home/test/GaN/dec/Intrinsic_Defect/V_N1+V_N1/nn1/initial_structure/q0
Job 119428 submitted: /data/home/test/GaN/dec/Intrinsic_Defect/V_N1+V_N1/nn3/initial_structure/q0
Job 119430 submitted: /data/home/test/GaN/dec/Intrinsic_Defect/Ga_N1+N_Ga1/nn2/initial_structure/q0
Job 119432 submitted: /data/home/test/GaN/dec/Intrinsic_Defect/Ga_N1+N_Ga1/nn1/initial_structure/q0
Job 119434 submitted: /data/home/test/GaN/dec/Intrinsic_Defect/Ga_N1+N_Ga1/nn3/initial_structure/q0
...
############ AutoRun module end ############
至此电中性复合缺陷的结构优化及总能计算完成。
5.8.4.3. GaN带电复合缺陷结构优化及总能计算流程¶
检查GaN中带电缺陷的结构优化是否完成
如果带电点缺陷的结构优化及总能计算已经完成,DASP则会自动跳过
############ Ionized Defect module start ############
Ionized defect exist at: /data/home/test/GaN/dec/Intrinsic_Defect/V_N1/initial_structure, skipped
Ionized defect exist at: /data/home/test/GaN/dec/Intrinsic_Defect/N_Ga1/initial_structure, skipped
...
GaN带电复合缺陷结构产生
DASP会根据中性复合缺陷的计算结果判断可能的带电状态并完成带电复合缺陷的结构优化及总能的计算:
Start generating ionized defects
Ionized defect path: /data/home/test/GaN/dec/Intrinsic_Defect/V_N1+V_N1/nn2/initial_structure/q+1
Ionized defect path: /data/home/test/GaN/dec/Intrinsic_Defect/V_N1+V_N1/nn2/initial_structure/q+2
Ionized defects generation completed
Start generating ionized defects
Ionized defect path: /data/home/test/GaN/dec/Intrinsic_Defect/V_N1+V_N1/nn1/initial_structure/q+1
Ionized defect path: /data/home/test/GaN/dec/Intrinsic_Defect/V_N1+V_N1/nn1/initial_structure/q+2
Ionized defects generation completed
Start generating ionized defects
Ionized defect path: /data/home/test/GaN/dec/Intrinsic_Defect/V_N1+V_N1/nn3/initial_structure/q+1
Ionized defect path: /data/home/test/GaN/dec/Intrinsic_Defect/V_N1+V_N1/nn3/initial_structure/q+2
Ionized defects generation completed
Start generating ionized defects
...
提交任务优化带电复合缺陷的结构并完成总能的计算
############ AutoRun module start ############
Job 119494 submitted: /data/home/test/GaN/dec/Intrinsic_Defect/V_N1+V_N1/nn2/initial_structure/q+2
Job 119496 submitted: /data/home/test/GaN/dec/Intrinsic_Defect/V_N1+V_N1/nn2/initial_structure/q+1
Job 119498 submitted: /data/home/test/GaN/dec/Intrinsic_Defect/V_N1+V_N1/nn1/initial_structure/q+2
Job 119500 submitted: /data/home/test/GaN/dec/Intrinsic_Defect/V_N1+V_N1/nn1/initial_structure/q+1
Job 119502 submitted: /data/home/test/GaN/dec/Intrinsic_Defect/V_N1+V_N1/nn3/initial_structure/q+2
Job 119504 submitted: /data/home/test/GaN/dec/Intrinsic_Defect/V_N1+V_N1/nn3/initial_structure/q+1
...
5.8.4.4. GaN复合缺陷形成能计算及绘图¶
GaN复合缺陷形成能计算
优化完各个价态结构并计算得到总能后,DASP会计算所有缺陷(包括点缺陷)各个价态的形成能及转变能级。
############ Formation Energy module start ############
Start the formation energy calculation
The formation energy (neutral) of V_N1 at p1 is 3.169 eV
The formation energy (neutral) of V_N1 at p2 is 4.474 eV
The FNV correction (q = 2) E_correct = 0.332 eV
The transition level (0/2+) above VBM: 1.658 eV
The FNV correction (q = 1) E_correct = 0.136 eV
The transition level (0/+) above VBM: 3.466 eV
The FNV correction (q = 3) E_correct = 1.206 eV
The transition level (0/3+) above VBM: 1.601 eV
The formation energy calculation completed
...
############ Formation Energy module end ############
GaN复合缺陷形成能绘图
完成形成能的计算后,DASP会绘制所有缺陷(包括点缺陷,复合缺陷)在特定化学势条件下的形成能随费米能级变化图。
############ Plot Diagram module start ############
Start plotting the diagrams
Generate formation energy diagrams at p1: /data/home/test/GaN/dec/Formation_Energy_Intrinsic_Defect/p1.dat
Generate formation energy diagrams at p2: /data/home/test/GaN/dec/Formation_Energy_Intrinsic_Defect/p2.dat
Generate transition level diagram: /data/home/test/GaN/dec/Transition_Level_Intrinsic_Defect/tl.dat
All diagrams completed
Possible defect complexes:
1 V_N1+[V_N1+V_N1]-3
2 V_N1+[V_N1+V_N1]-1
3 V_N1+[V_Ga1+V_N1]-1
4 V_Ga1+V_N1
5 V_Ga1+[V_Ga1+V_N1]-1
############ Plot Diagram module end ############
DASP会绘制所有缺陷的形成能随费米能级的变化图,此处仅显示了所考虑的复合缺陷及与之相关的点缺陷形成能随费米能级变化图。
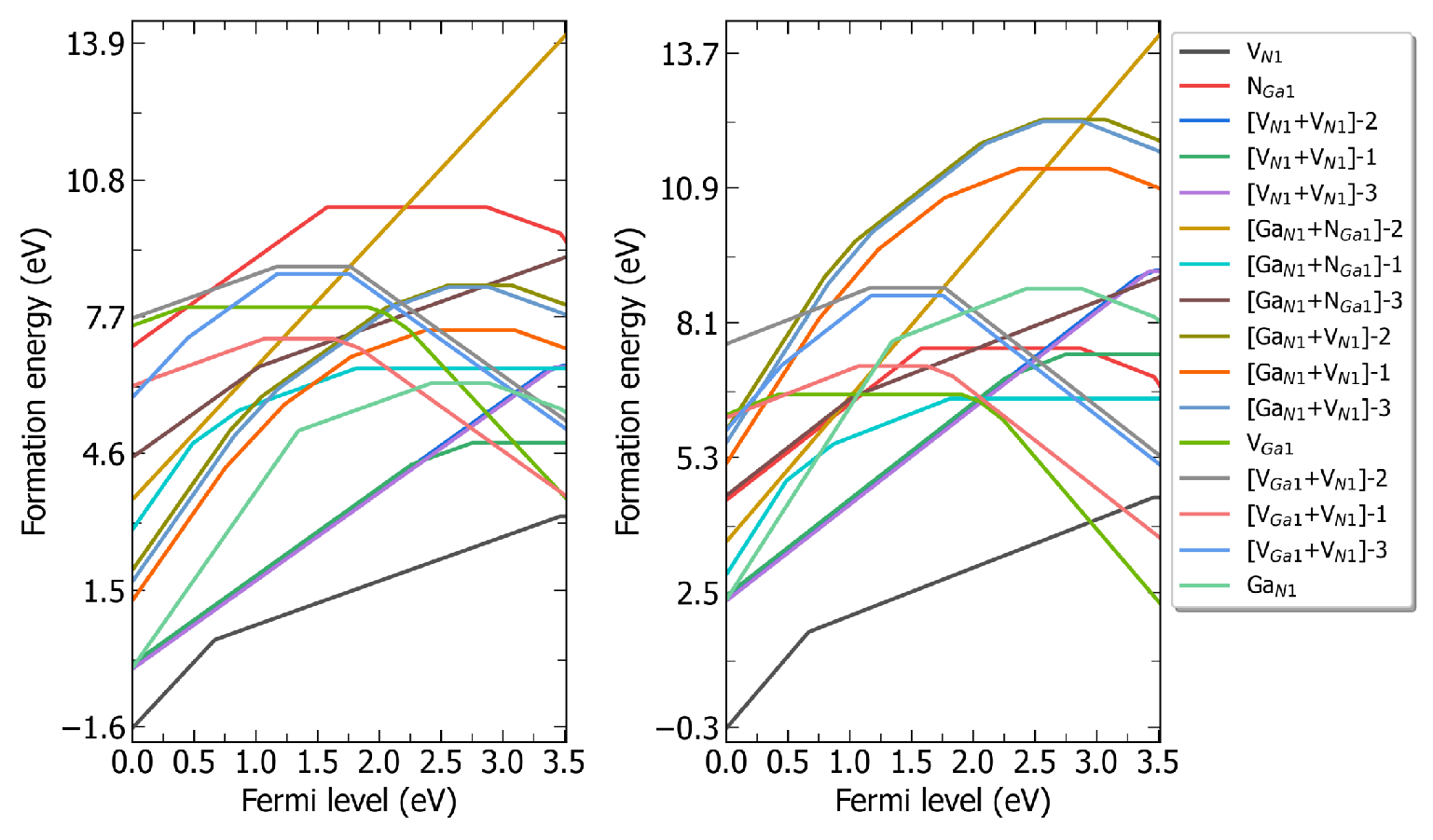
GaN中复合缺陷及对应点缺陷在化学势点(a) p1, (b) p2处的形成能随费米能级的变化。
从图中可以发现:对于这四种复合缺陷,总是最近邻构型(即 nn1 )的形成能更低。
至此DASP已经完成了用户设置的所有缺陷的形成能、转变能级的计算,如果要完成缺陷(杂质)浓度及载流子动力学的计算,继续执行 dasp 4 以及 dasp 5 即可。