1. 软件简介¶
1.1. 功能介绍¶
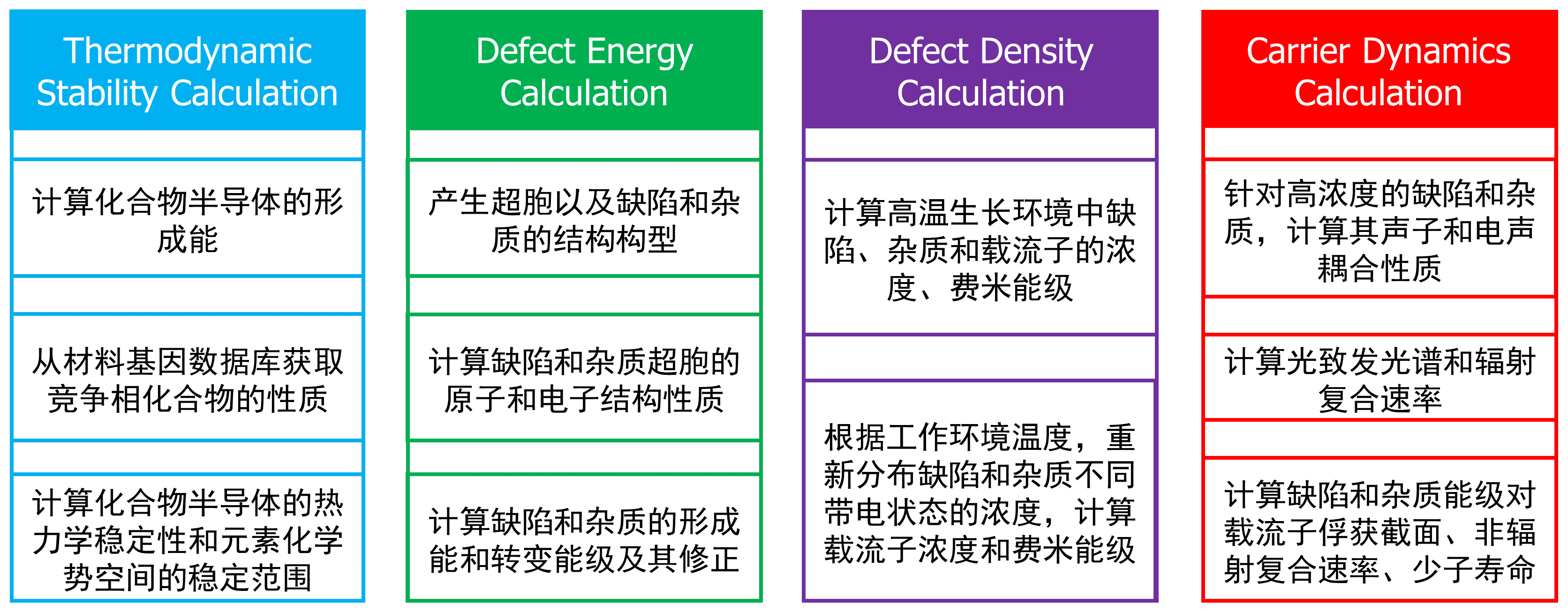
DASP的TSC,DEC,DDC和CDC模块的功能介绍。
1.2. 计算流程¶
POSCAR 和DASP计算参数文件 dasp.in 。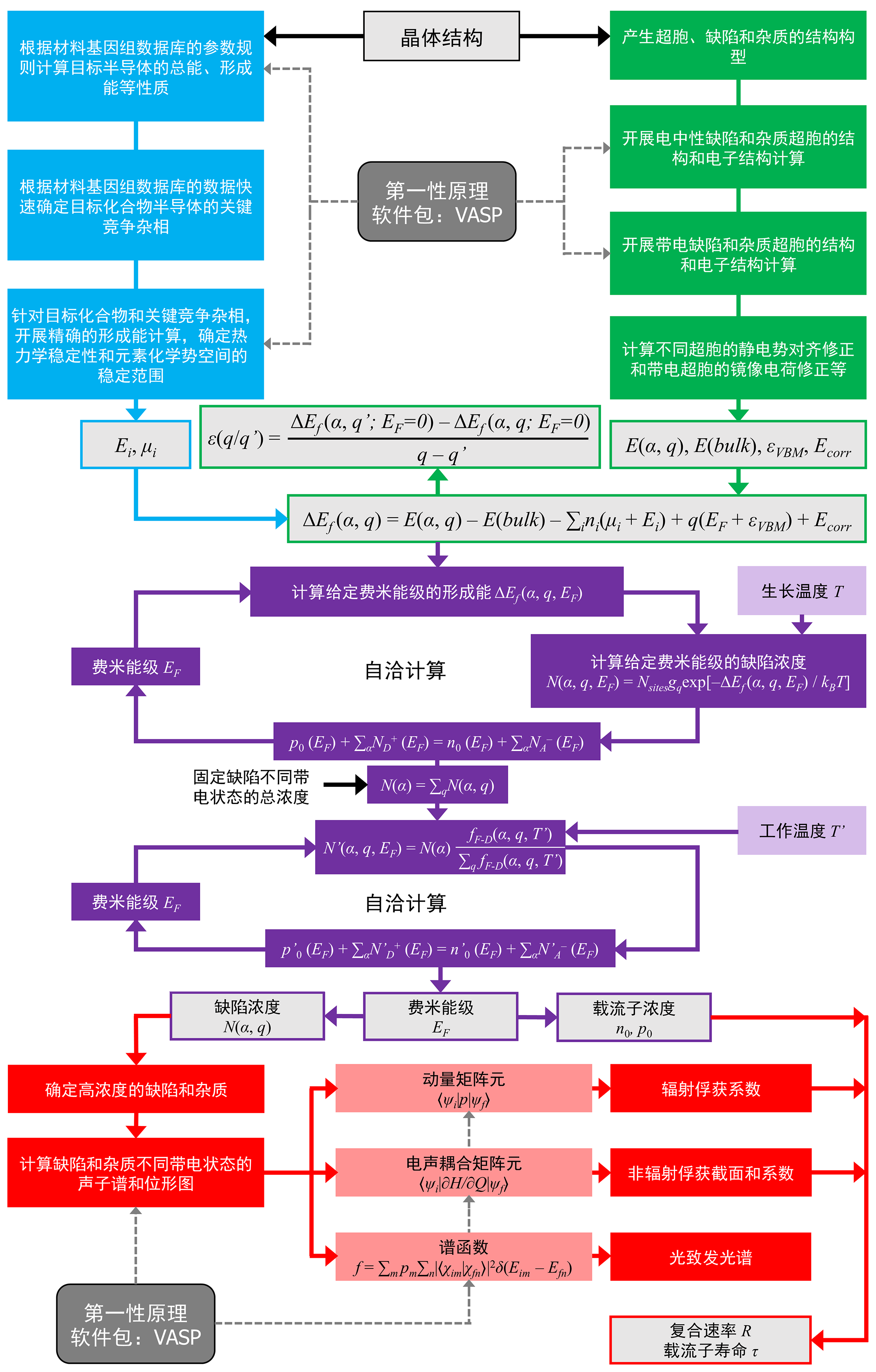
DASP的计算流程图。
POSCAR 和 dasp.in 这两个输入文件后,依次执行以下5条命令,即可完成所有计算:dasp 1 (对应PREPARE)dasp 2 (对应TSC)dasp 3 (对应DEC)dasp 4 (对应DDC)dasp 5 (对应CDC)tsc-state 查询计算任务进展。在DEC计算时,可以通过 dec-state 查询计算任务进展。1.3. 运行环境需求¶
1.3.1. 第一性原理计算软件:VASP¶
DASP软件需调用第一性原理计算软件包Vienna Ab initio Simulation Package (VASP)来开展缺陷相关的结构和电子结构计算,因此,用户需要提供已经编译好的VASP可执行文件目录。
1.3.2. Materials Project与本地数据库¶
pip install 命令自动安装Pymatgen与mp-api。Materials Project 在线数据库。若选择访问 Materials Project 在线数据库,用户须在 Materials Project 注册账号并获取API Key。备注
After installation, do:
pmg config -p <EXTRACTED_VASP_POTCAR> <MY_PSP>
In the above, <EXTRACTED_VASP_POTCAR> is the location of the directory that you extracted the downloaded VASP pseudopotential files. Typically, it has the following format:
- <EXTRACTED_VASP_POTCAR>
|- POT_GGA_PAW_PBE #(此处必须为2003版)
||- Ac_s
|||-POTCAR
|||-...
or
- <EXTRACTED_VASP_POTCAR>
|- potpaw_PBE #(此处必须为2003版)
||- Ac_s
|||-POTCAR
|||-...
and follow the instructions. If you have done it correctly, you should get a resources directory with the following directory structure:
- psp_resources
|- POT_GGA_PAW_PBE
||- POTCAR.Ac_s.gz
||- POTCAR.Ac.gz
||- POTCAR.Ag.gz
...
|- POT_GGA_PAW_PW91
...
After generating the resources directory, you should add a VASP_PSP_DIR config variable pointing to the generated directory and you should then be able to generate POTCARs:
pmg config --add PMG_VASP_PSP_DIR <MY_PSP>
1.4. 使用范围¶
DASP软件能计算半导体和绝缘体的缺陷和杂质性质,因此,用户在计算之前需要通过能带计算检查目标材料是否有带隙。对于部分窄带隙半导体,GGA和LDA等交换关联近似可能造成带隙低估甚至消失,此时需要采用杂化泛函等开展计算(详见具体参数部分的level=2和3)。
- 1
Menglin Huang, Zhengneng Zheng, Zhenxing Dai, Xinjing Guo, Shanshan Wang, Lilai Jiang, Jinchen Wei, and Shiyou Chen. Dasp: defect and dopant ab-initio simulation package. Journal of Semiconductors, 43(4):042101, 2022. doi:10.1088/1674-4926/43/4/042101.
- 2
Menglin Huang, Zenghua Cai, Shanshan Wang, Xin-Gao Gong, Su-Huai Wei, and Shiyou Chen. More se vacancies in sb2se3 under se-rich conditions: an abnormal behavior induced by defect-correlation in compensated compound semiconductors. Small, 17(36):2102429, 2021. doi:10.1002/smll.202102429.
- 3
Menglin Huang, Shan-Shan Wang, Yu-Ning Wu, and Shiyou Chen. Defect physics of ternary semiconductor zn ge p 2 with a high density of anion-cation antisites: a first-principles study. Physical Review Applied, 15(2):024035, 2021. doi:10.1103/PHYSREVAPPLIED.15.024035.
- 4
Jinchen Wei, Lilai Jiang, Menglin Huang, Yuning Wu, and Shiyou Chen. Intrinsic defect limit to the growth of orthorhombic hfo2 and (hf, zr) o2 with strong ferroelectricity: first-principles insights. Advanced Functional Materials, 31(42):2104913, 2021. doi:10.1002/adfm.202104913.
- 5
Shiyou Chen, Aron Walsh, Xin-Gao Gong, and Su-Huai Wei. Classification of lattice defects in the kesterite cu2znsns4 and cu2znsnse4 earth-abundant solar cell absorbers. Advanced materials, 25(11):1522–1539, 2013. doi:10.1002/adma.201203146.