1 Introduction¶
1.1 What is DASP¶
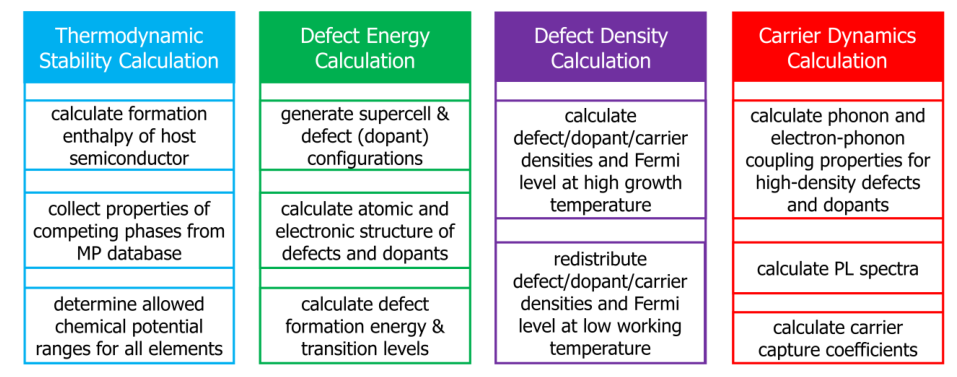
Fig 1. The framework of the DASP software, which is composed of four modules, TSC, DEC, DDC and CDC. The major functions of the four modules are shown in the boxes.
1.2 Calculation flow¶
POSCAR and dasp.in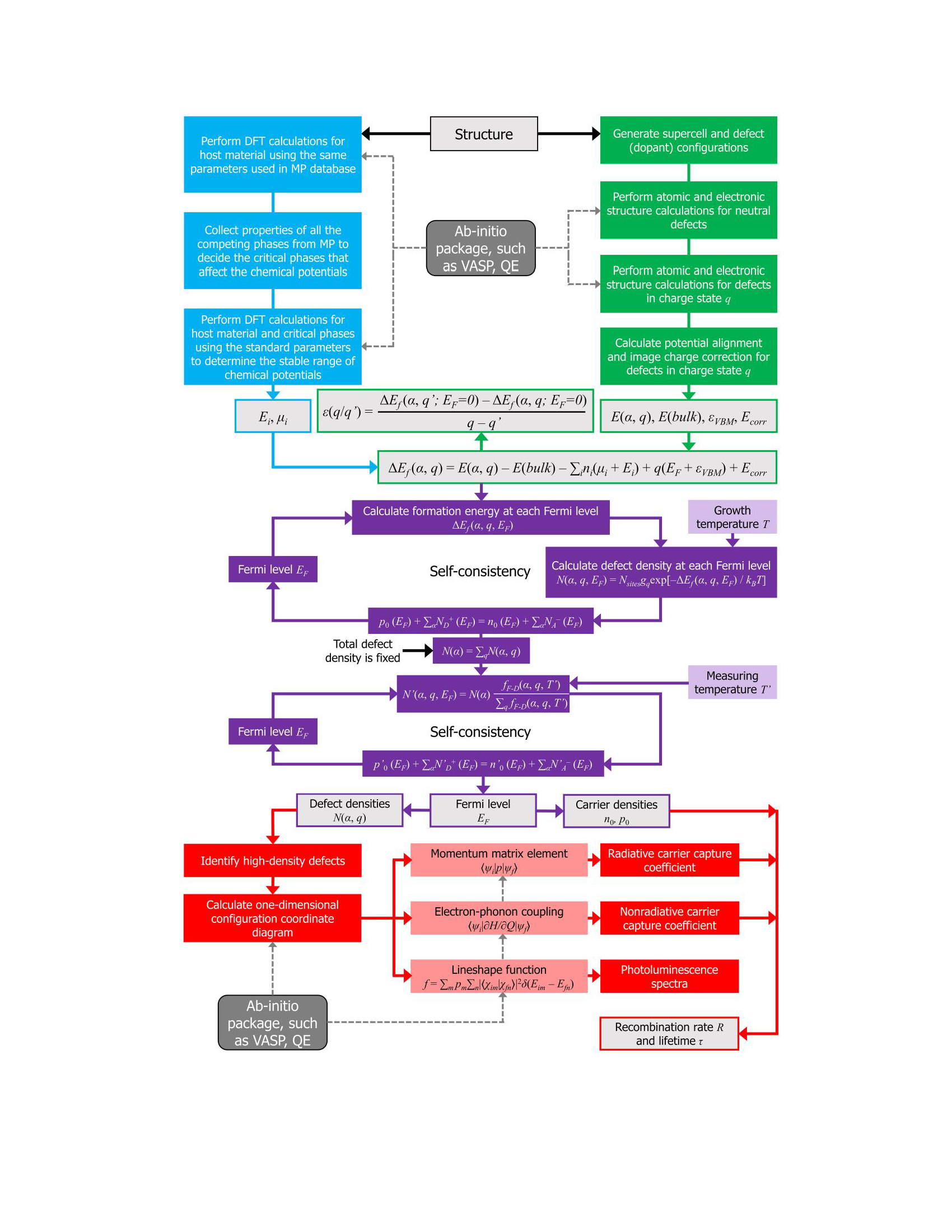
Fig 2. The flowchart of DASP. Different colors represent the four modules. The dashed lines show the calculations that need to call external ab-initio DFT softwares.
POSCAR and dasp.in :dasp 1 (corresponding to PREPARE)dasp 2 (corresponding to TSC)dasp 3 (corresponding to DEC)dasp 4 (corresponding to DDC)dasp 5 (corresponding to CDC)tsc-state and dec-state during TSC and DEC calculation, respectively.1.3 Operating environment requirements¶
1.3.1 First-principle calculation software: VASP¶
The first-principles calculation software package, Vienna Ab initio Simulation Package (VASP), is needed to perform defect-related structural and electronic structure calculations for the DASP software. Therefore, the user needs to provide the compiled VASP executable file directory.
1.3.2 Materials Project database¶
pip install command.Materials Project database will be accessed through Pymatgen, so the user must register an account in Materials Project and obtain the API.After installation, do:
pmg config -p <EXTRACTED_VASP_POTCAR> <MY_PSP>
In the above, <EXTRACTED_VASP_POTCAR> is the location of the directory that you extracted the downloaded VASP pseudopotential files. Typically, it has the following format:
- <EXTRACTED_VASP_POTCAR>
|- POT_GGA_PAW_PBE #(must be version 2003)
||- Ac_s
|||-POTCAR
|||-...
or
- <EXTRACTED_VASP_POTCAR>
|- potpaw_PBE #(must be version 2003)
||- Ac_s
|||-POTCAR
|||-...
and follow the instructions. If you have done it correctly, you should get a resources directory with the following directory structure:
- psp_resources
|- POT_GGA_PAW_PBE
||- POTCAR.Ac_s.gz
||- POTCAR.Ac.gz
||- POTCAR.Ag.gz
...
|- POT_GGA_PAW_PW91
...
After generating the resources directory, you should add a VASP_PSP_DIR config variable pointing to the generated directory and you should then be able to generate POTCARs:
pmg config --add PMG_VASP_PSP_DIR <MY_PSP>
1.4 Scope of use¶
DASP can calculate the defect and impurity properties of semiconductors and insulators. Therefore, users need to check whether the target material has a band gap through the band structure before defect calculation. For some narrow-bandgap semiconductors, exchange-correlation approximations such as GGA and LDA may cause the bandgap to be underestimated or even disappear. In this case, hybrid functionals are required for calculation (see level=2 and 3 in the specific parameters section for details).