10. 科学计算软件应用实例¶
10.1. Nanodcal 实例¶
Nanodcal 是一款基于非平衡格林函数-密度泛函理论(NEGF-DFT)的第一性原理计算软件,主要用于模拟器件材料中的非线性、非平衡的量子输运过程,是目前国内唯一一款拥有自主知识产权的基于第一性原理的输运软件。可预测材料的电流-电压特性、电子透射几率等众多输运性质。
迄今为止,Nanodcal 已成功应用于1维、2维、3维材料物性、分子电子器件、自旋电子器件、光电流器件、半导体电子器件设计等重要研究课题中,并将逐步推广到更广阔的电子输运性质研究的领域。
以 Benzene分子的自洽、本征态计算 为例详细描述 Nanodcal 在Device Studio中的应用。包含创建项目,从建模到计算,到数据的可视化分析整个流程。Device Studio可以生成 Nanodcal 很多功能计算的输入文件,用户可根据计算需要选择生成。
10.1.1. Nanodcal计算流程¶


10.1.2. Nanodcal创建项目¶
双击Device Studio图标快捷方式,登录并启动Device Studio,在创建或打开项目界面中(启动软件后选择创建或打开项目的图形界面),根据界面提示选择创建一个新的项目( Create a new Project )或打开一个已经存在的项目( Open an existing Project )的按钮,选中之后点击界面中的 OK 按钮即可。若选择创建一个新的项目,用户可根据需要给该项目命名,如本项目命名为 Nanodcal,或采用软件默认项目名。
10.1.3. Nanodcal搭建或导入结构¶
对于想要搭建的结构,用户可先在Device Studio本地数据库或在线数据库搜索结构是否存在。若存在,找到结构直接导入;若不存在,可将库中的相关结构先导入,在此基础上方便快捷的搭建,也可直接自行搭建。如Benzene分子,该结构在Device Studio本地库中存在,则可在本地库中找到该结构直接导入即可,导入Benzene分子结构后的图形界面如 图 10.3 所示。在Device Studio中导入Benzene分子结构操作这里不做详细说明,用户可参照 导入结构 节内容。
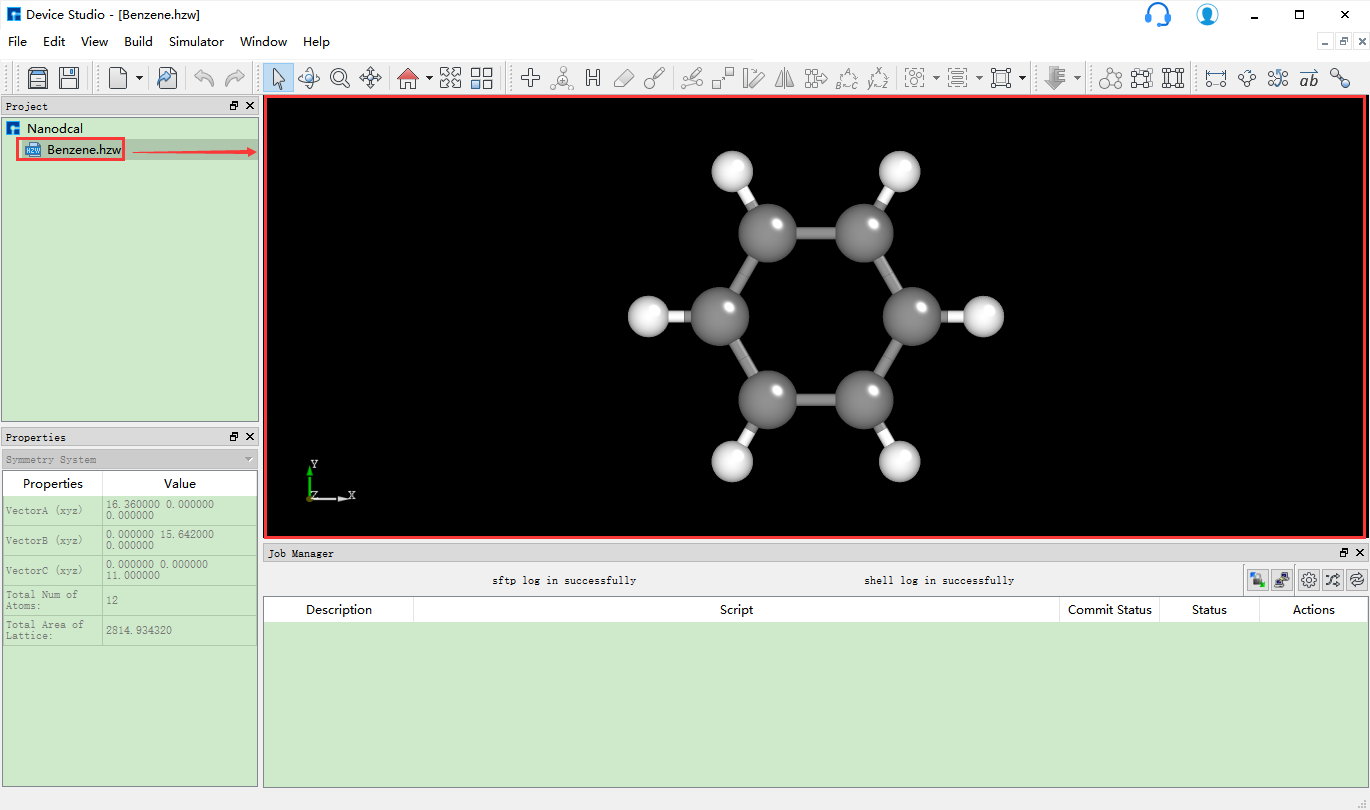
图 10.3 导入Benzene分子结构后的图形界面¶
10.1.4. Nanodcal输入文件的生成¶
10.1.4.1. Nanodcal生成自洽计算输入文件¶
在如 图 10.3 所示界面中选中 Simulator → Nanodcal → SCF Calculation,弹出界面如 图 10.4 所示,用户可根据所计算的结构及计算需要分别选中 Basic settings 、Iteration control、Basis set、Spin type 这四个按钮合理设置参数,之后点击 Generate files 即可生成自洽计算输入文件。
如生成Benzene分子自洽计算输入文件,根据计算需要设置参数,分别选中 Basic settings 、Iteration control、Basis set、Spin type 设置参数分别如 图 10.4 、 图 10.5 、 图 10.6 和 图 10.7 所示,设置好参数后点击 Generate files 即可生成Benzene自洽计算输入文件 scf.input。
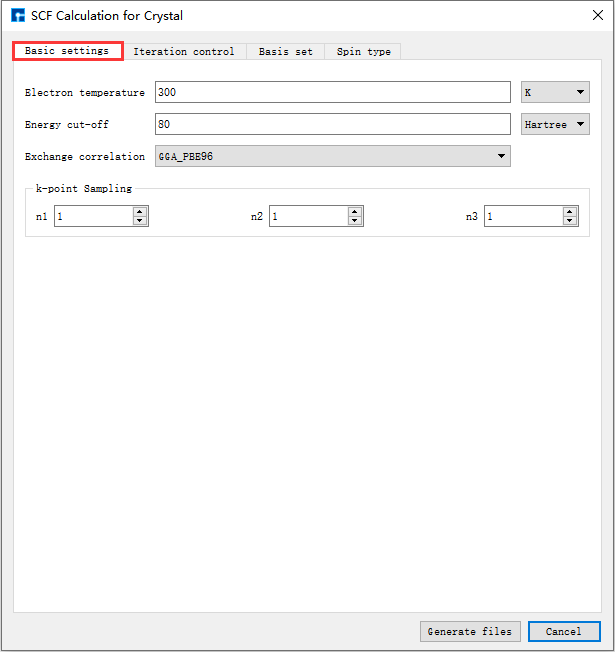
图 10.4 Basic settings参数设置界面¶
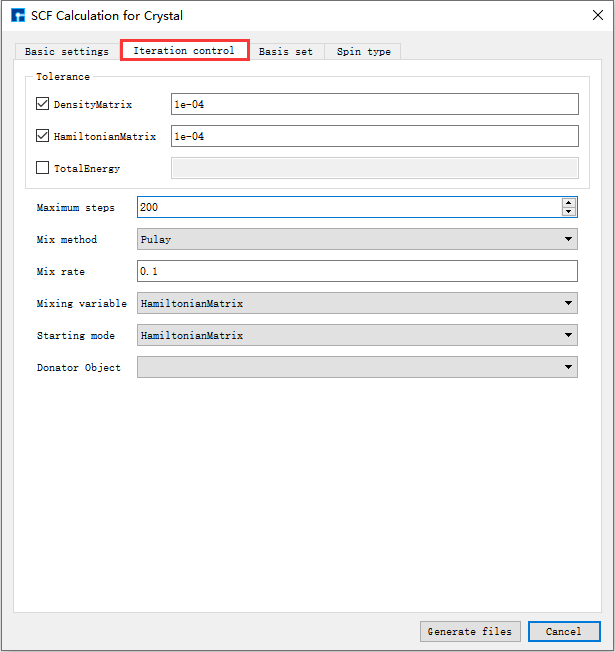
图 10.5 Iteration control参数设置界面¶
图 10.6 Basis set参数设置界面¶
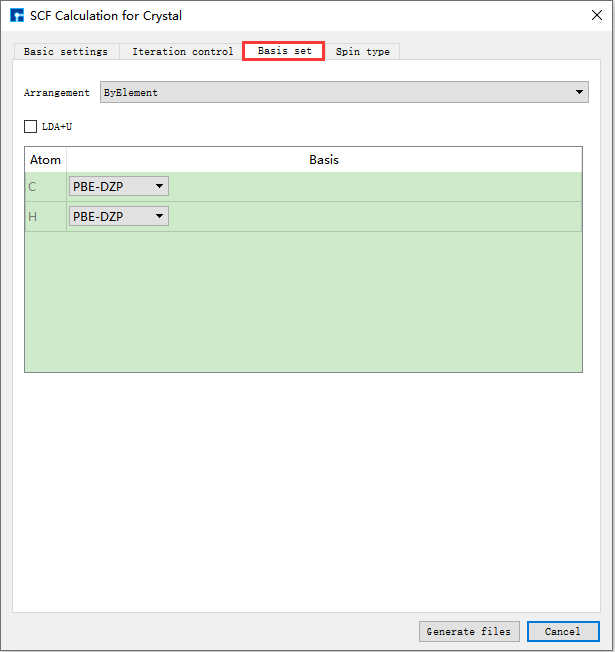
图 10.7 Spin type参数设置界面¶
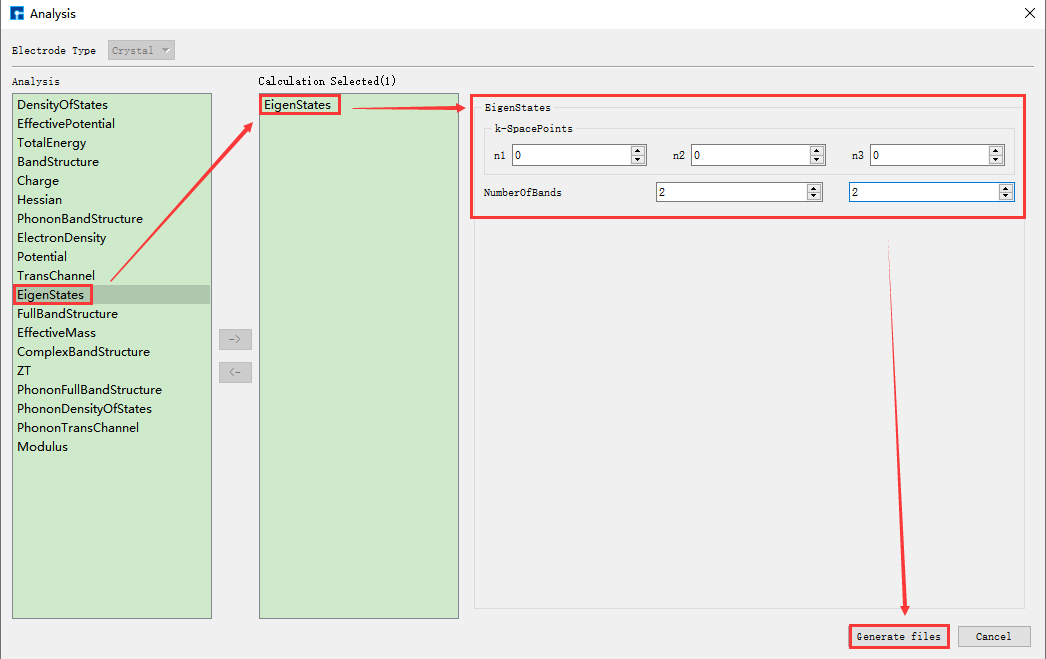
10.1.5. Nanodcal连接服务器¶
生成输入文件之后需要计算,计算之前需连接装有Nanodcal的服务器。如连接装有Nanodcal的 内蒙A区 超算服务器,则可在如 图 10.9 所示界面中点击 设置 按钮,弹出MachineOptions界面,在MachineOptions界面中点击 内蒙A区 ,点击 Select 则连接好装有Nanodcal的 内蒙A区 超算服务器。同理,若要连接装有Nanodcal的本地电脑,则在 图 10.9 中所示的MachineOptions界面上选中MyComputer,点击 Select 即可。
若用户想要添加新的装有Nanodcal的服务器,在如 图 10.10 所示界面中点击 设置 按钮,弹出MachineOptions界面,在该界面中点击 New 按钮,弹出MachineSet界面,在该界面中填写Computer Name、HostIp、Port、Username、Password等一系列信息,点击 OK 按钮则在MachineOptions界面中添加了装有Nanodcal的服务器。
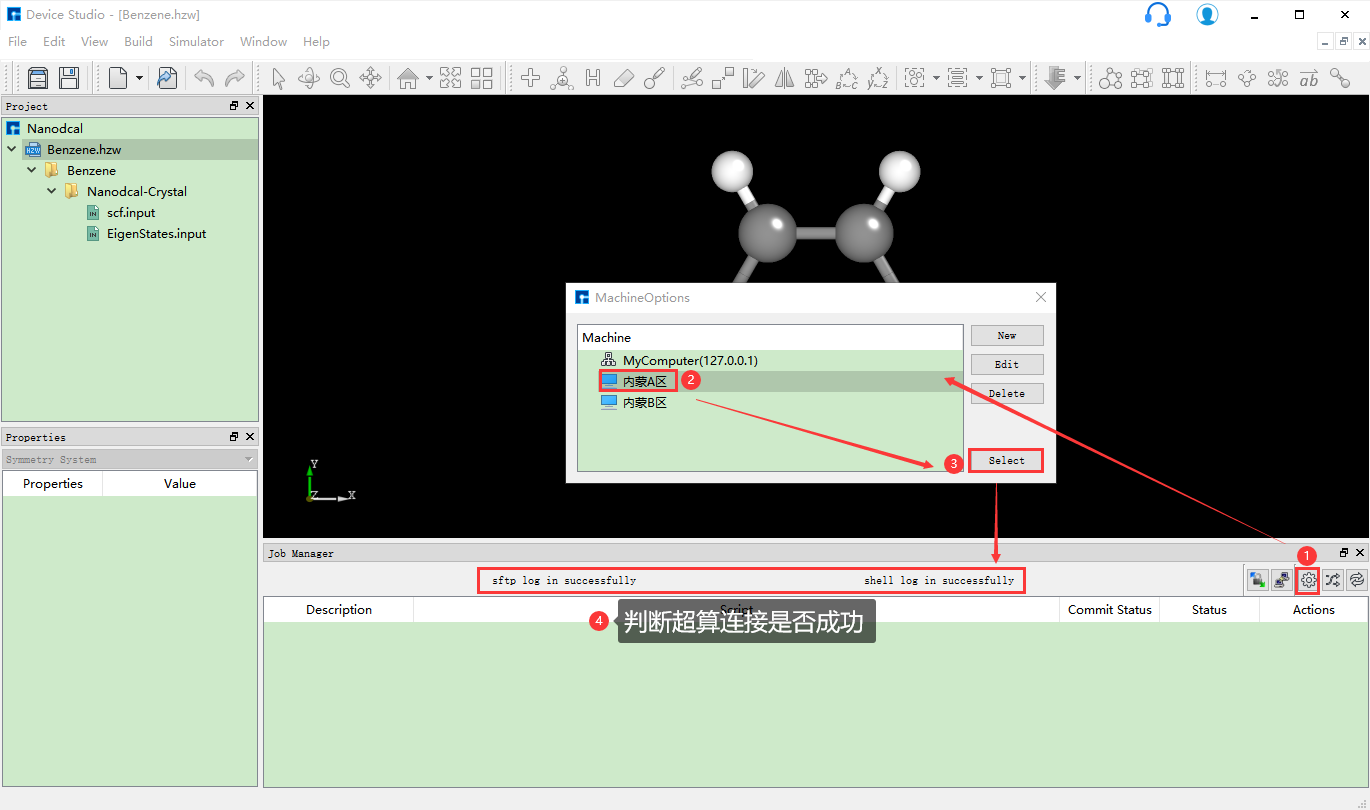
图 10.9 连接服务器操作界面¶
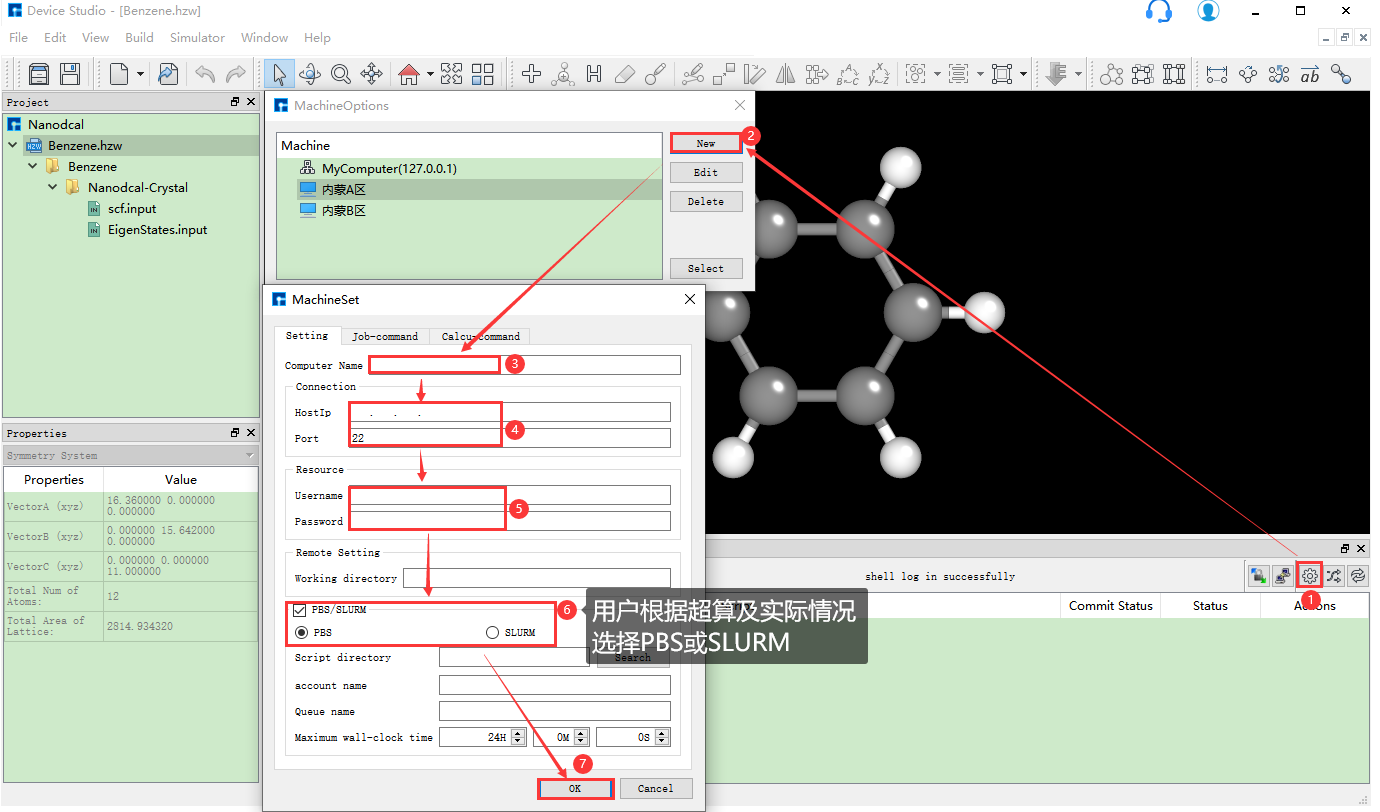
图 10.10 添加并连接新的超算服务器操作界面¶
10.1.6. Nanodcal计算¶
10.1.6.1. Nanodcal自洽计算¶
在做自洽计算之前需查看计算所需的输入文件(scf.input)和基组文件(C_PBE-DZP.nad 和 H_PBE-DZP.nad)是否齐全,再根据计算需要打开 scf.input 文件并查看文件中的参数设置是否合理,若不合理,则可选择直接在文件中进行编辑或重新生成,最后再进行自洽计算。具体步骤如下:
(1)连接装有Nanodcal的服务器。
(2)检查自洽计算所需的输入文件(scf.input)和基组文件(C_PBE-DZP.nad 和 H_PBE-DZP.nad)是否齐全。选中 scf.input → 右击 → Open with 即可查看到Benzene分子自洽计算输入文件如下所示:
%%What quantities should be calculated
calculation.name = scf
%Basic setting
calculation.occupationFunction.temperature = 300
calculation.realspacegrids.E_cutoff = 80 Hartree
calculation.xcFunctional.Type = GGA_PBE96
calculation.k_spacegrids.number = [ 1 1 1 ]'
system.centralCellVectors = [[16.36 0 0]' [0 15.642 0]' [0 0 11]']
system.spinType = NoSpin
%Iteration control
calculation.SCF.monitoredVariableName = {'rhoMatrix','hMatrix','totalEnergy','bandEnergy','gridCharge','orbitalCharge'}
calculation.SCF.convergenceCriteria = {1e-04,1e-04,[],[],[],[]}
calculation.SCF.maximumSteps = 200
calculation.SCF.mixMethod = Pulay
calculation.SCF.mixRate = 0.1
calculation.SCF.mixingMode = H
calculation.SCF.startingMode = H
%calculation.SCF.donatorObject = NanodcalObject.mat
%Basic set
system.neutralAtomDataDirectory = '../'
system.atomBlock = 12
AtomType OrbitalType X Y Z
C PBE-DZP 5.00900000 5.57200000 5.00000000
C PBE-DZP 5.77900000 4.23800000 5.00000000
C PBE-DZP 7.31900000 4.23800000 5.00000000
C PBE-DZP 8.08900000 5.57200000 5.00000000
C PBE-DZP 7.31900000 6.90500000 5.00000000
C PBE-DZP 5.77900000 6.90500000 5.00000000
H PBE-DZP 3.86900000 5.57200000 5.00000000
H PBE-DZP 5.20900000 3.25100000 5.00000000
H PBE-DZP 7.88900000 3.25100000 5.00000000
H PBE-DZP 9.22900000 5.57100000 5.00000000
H PBE-DZP 7.88900000 7.89200000 5.00000000
H PBE-DZP 5.21000000 7.89300000 5.00000000
end
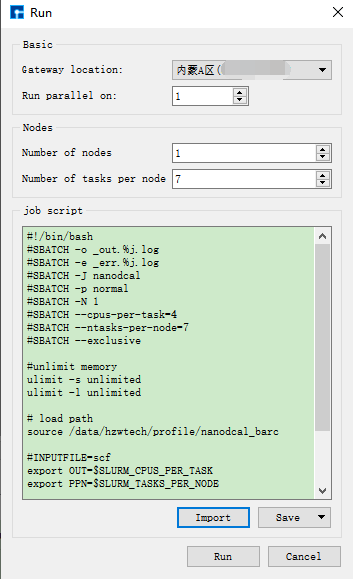
图 10.11 Run界面¶
(3)在Device Studio的Project Explorer区域选中 scf.input → 右击 → Run,弹出Run界面如 图 10.11 所示。根据计算需要设置参数后点击 Save 按钮保存相应的脚本,之后点击如 图 10.11 所示界面中的 Run 按钮则可做Benzene分子的自洽计算。用户可在Job Manager区域中观察自洽计算状态,当自洽计算任务处于排队中、计算中和计算完成时,Status 分别为Queued、Running、Finished;在计算过程中,用户可根据需要点击 刷新 按钮,通过 Commit Status 、Status 可查看到自洽计算的计算进度和计算状态;计算完成后Device Studio的Job Manager区域如 图 10.12 所示,点击 Action 下的 下载 按钮弹出下载Benzene分子自洽计算结果界面如 图 10.13 所示,在该界面找到自洽计算的结果文件 NanodcalObject.mat,点击 下载 按钮则可下载,下载后可在软件的Project Explorer区域查看到该结果文件。
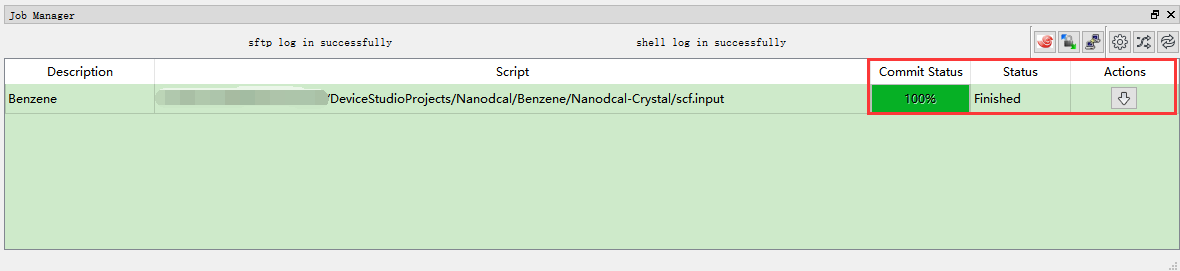
图 10.12 Benzene分子自洽计算完成的Device Studio的Job Manager区域¶
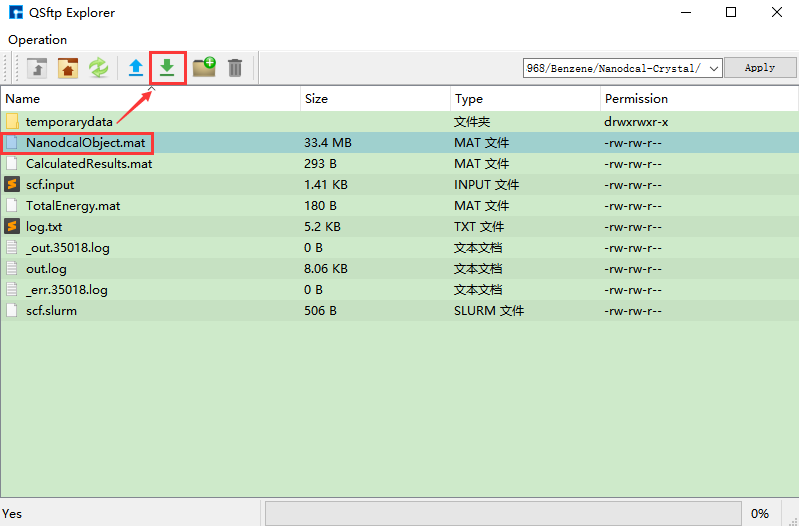
图 10.13 下载Benzene分子自洽计算结果界面¶
10.1.6.2. Nanodcal本征态计算¶
本征态计算需调用自洽计算结果,故需先做自洽计算,再做本征态计算。本算例已经做过自洽计算,则不需要再做自洽计算,直接调用自洽计算结果做本征态计算即可。Benzene分子的本征态计算的具体步骤如下:
(1)连接装有Nanodcal的服务器。
(2)选中 EigenStates.input → 右击 → Open with 即可查看到Benzene分子本征态计算输入文件如下所示:
system.object = NanodcalObject.mat
calculation.name = eigenStates
calculation.eigenStates.kSpacePoints = [0 0 0]'
calculation.eigenStates.numberOfBands = [2,2]
calculation.eigenStates.realSpace = true
calculation.eigenStates.plot = true
%calculation.control.xml = true
calculation.control.dsf = 1
(3)在Device Studio的Project Explorer区域选中 EigenStates.input → 右击 → Run,弹出Run界面如 图 10.11 所示。根据计算需要设置参数后点击 Save 按钮保存相应的脚本,点击界面中的 Run 按钮则可做Benzene分子的本征态计算,计算完成后下载本征态计算结果文件 EigenStates.dsf。
10.1.7. Nanodcal本征态的可视化分析¶
对于本征态的可视化分析有3种情况,分别为3维(3D)、2维(2D)和1维(1D),3种可视化分析可自由切换,其中2D的可视化分析又分为平面显示和立体显示2种情况,用户可根据需要进行选择。
10.1.7.1. 本征态的3D可视化分析¶
在Device Studio的Project Explorer区域选中本征态计算结果文件 EigenStates.dsf → 右击 → Show View,弹出本征态的3D可视化分析界面如 图 10.14 ,由 图 10.14 可知,用户可根据需要选择在可视化分析结果中是否显示该体系的结构信息以及显示该结构信息的方式;可选择是否显示Lattice框以及显示Lattice框的方式(分子结构没有Lattice框,故本算例中无法使用该功能);可通过改变Isovalue值来改变本征态3D可视化分析的形状;可通过选择如 图 10.14 中所示的红色框选部分来选择从不同平面来观察本征态的3D可视化分析结果;可通过滚动鼠标中键将3D可视化分析结果放大或缩小;可按住鼠标右键,通过拖动鼠标将3D可视化分析结果进行旋转;可根据需要选择不同的Colormap。在如 图 10.14 所示的界面中BandValues部分,用户可通过下拉选择显示第几条带的本征态可视化分析结果。
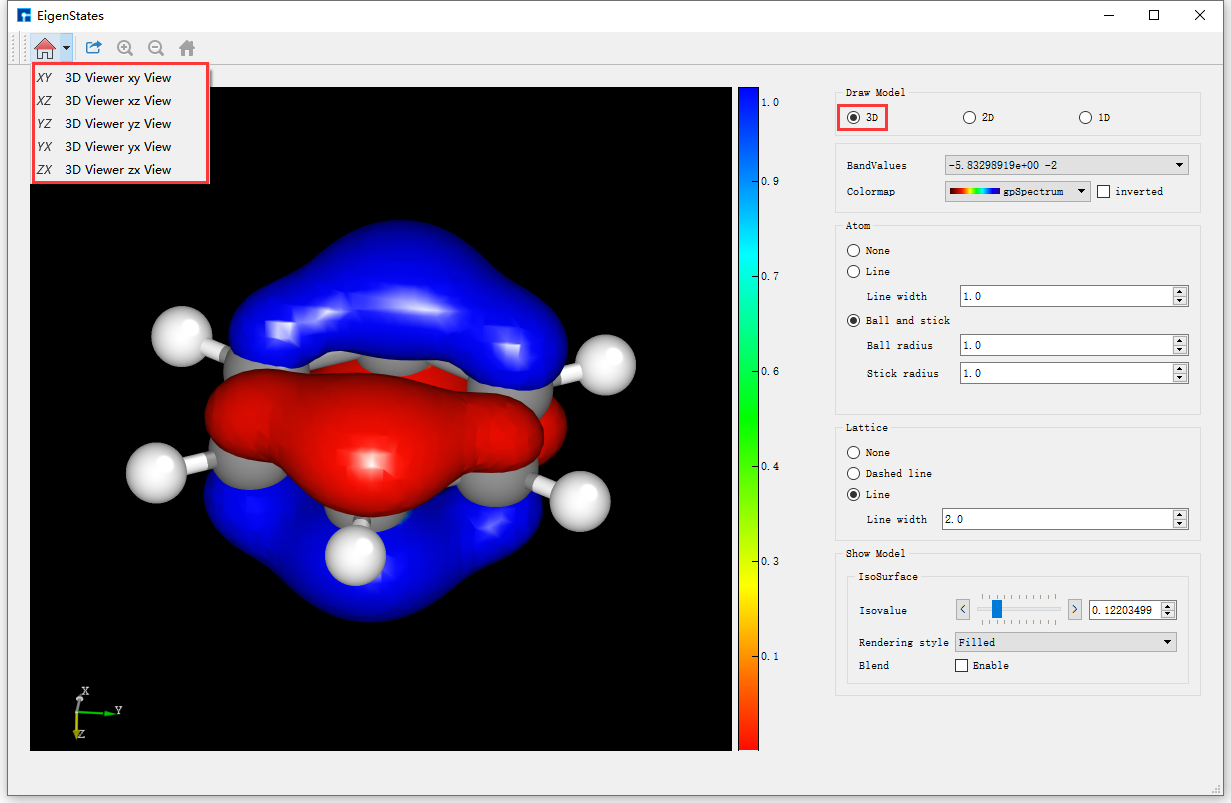
图 10.14 Benzene分子本征态的3D可视化分析界面¶
选中如 图 10.14 所示界面中 Export 快捷图标,弹出导出可视化分析结果的图形界面,用户可根据需要选择图片的保存路径和保存格式,并给所保存的图片命名。
对于不是通过Device Studio计算本征态,但含有本征态计算结果文件 EigenStates.dsf 的用户,可在如 图 10.3 所示界面中选中 Simulator → Nanodcal → Analysis Plot,弹出导入本征态结果文件的图形界面,在界面中选中本征态结果文件 EigenStates.dsf,点击 打开 按钮即可弹出本征态的3D可视化分析界面。
10.1.7.2. 本征态的2D可视化分析¶
图 10.14 右侧的Draw Model栏下的三个选项3D、2D和1D分别代表着计算结果的3D、2D和1D的可视化分析。若想对Benzene分子本征态计算结果进行2D的可视化分析,直接选中Draw Model栏下的2D按钮即可切换成本征态2D的可视化分析界面如 图 10.15 所示。
由 图 10.15 可知,2D的可视化分析分为两种情况,一种是平面显示(Plane display),一种是立体显示(Stereoscopic display),此刻选中的按钮为Plane display,即 图 10.15 亦为average along x的Benzene分子本征态2D平面显示的可视化分析结果。用户可根据需要通过选择Average direction栏下的3个按钮中的average along x、average along y或average along z来选择沿着x轴、y轴或z轴平均来查看并分析本征态的相关性质;可根据需要选择是否显示Colorbar,选择合适的Colormap,修改坐标轴的取值范围,修改标题和坐标轴的字体类型、字体大小以及字体是否加粗等。
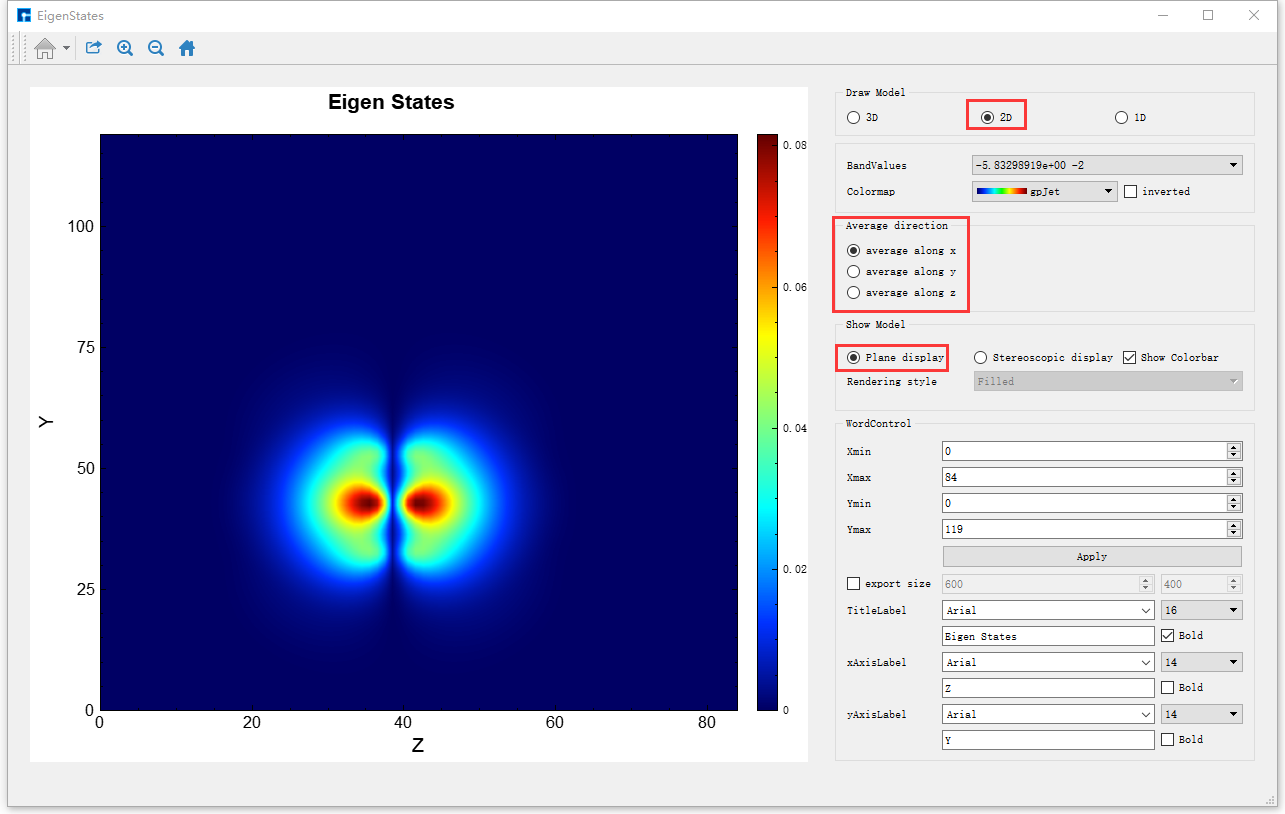
图 10.15 Benzene分子本征态的2D可视化分析界面(平面显示)¶
若想切换为立体显示,直接选中如 图 10.15 中的Stereoscopic display按钮即可,切换后的图形界面如 图 10.16 所示, 图 10.16 亦为average along x的Benzene分子本征态2D立体显示的可视化分析结果。对于如 图 10.16 图中左侧的图像,用户可通过滚动鼠标将图像放大或缩小;可通过拖动鼠标使图像发生任意角度和任意方向的旋转;可通过按住鼠标中键,拖动鼠标将图像进行平移。
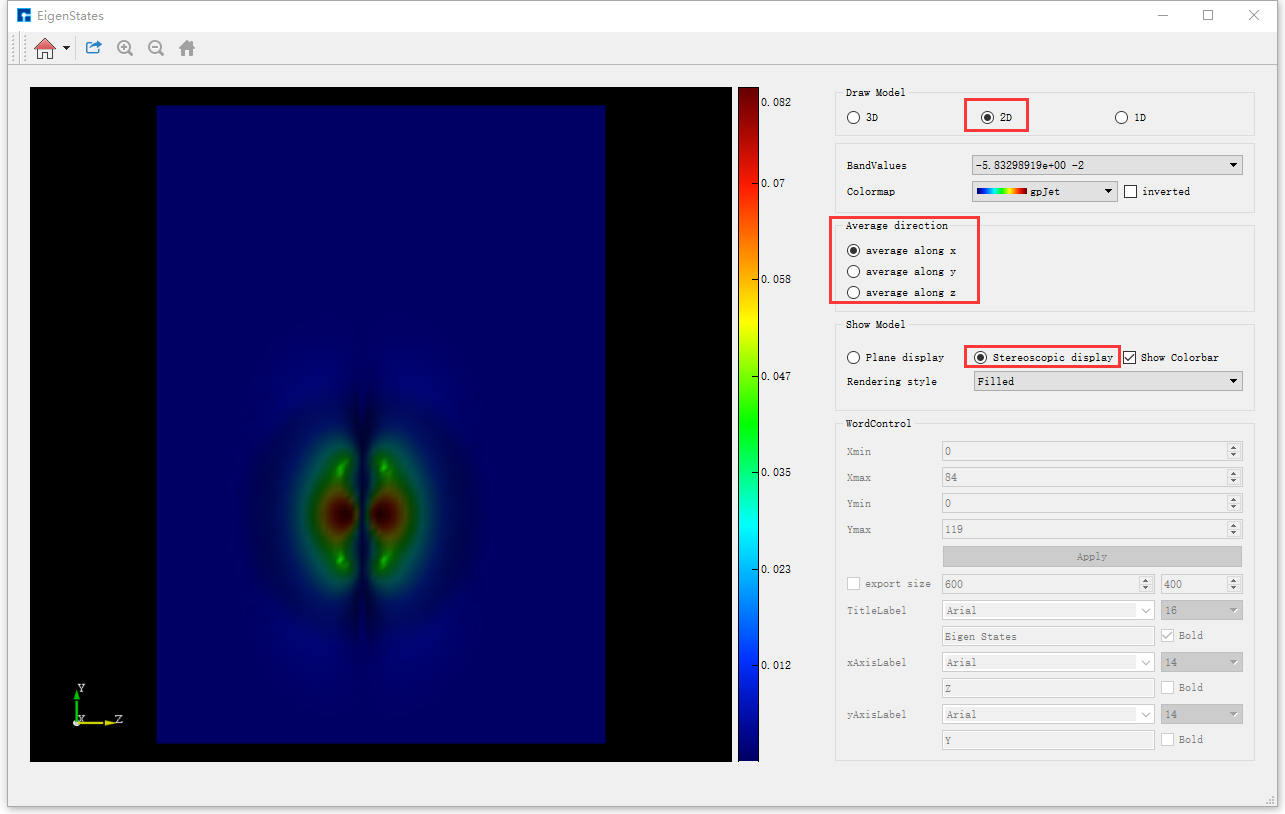
图 10.16 Benzene分子本征态的2D可视化分析界面(立体显示)¶
10.2. LAMMPS 实例¶
LAMMPS 即 Large-scale Atomic/Molecular Massively Parallel Simulator , 大规模原子分子并行模拟器 ,主要用于 分子动力学 相关的一些计算和模拟工作。一般来讲,分子动力学所涉及到的领域,LAMMPS代码也都涉及到了。LAMMPS由美国Sandia国家实验室开发,以GPL license发布,即开放源代码且可以免费获取使用,这意味着使用者可以根据自己的需要自行修改源代码。LAMMPS 程序在模拟固态材料(金属、半导体)、柔性物质(生物分子、聚合物)、粗粒度介观体系等方向具有广泛的应用。LAMMPS 程序内置多种原子间势(力场模型),可以实现原子、聚合物、生物分子、固态材料(金属、陶瓷、氧化物)、粗粒度体系的建模和模拟。该程序即可以模拟二维体系,也可以模拟三维体系,可以模拟多达数百万甚至数十亿粒子的分子体系,并提供支持多种势函数,具有良好的并行扩展性、模拟效率高、计算时间短等优点。
分子动力学模拟 ( Molecular Dynamics , MD )是近年来飞速发展的一种分子模拟方法,已经被广泛应用于化学化工、材料科学与工程、物理、生物医药等科学和技术领域,起到越来越重要的作用。MD模拟用来研究不能用解析方法来解决的复合体系的平衡性质和力学性质,用来搭建理论和实验的桥梁,在数学、生物、化学、物理学、材料科学和计算机科学交叉学科占据重要地位。
LAMMPS软件官网 :详见 https://lammps.sandia.gov
鸿之微科技(上海)股份有限公司在 Device Studio 2021B 中开发了适用于分子动力学计算软件 LAMMPS 的计算模块。使用Device Studio,用户可在其图形界面中方便快捷的搭建或导入计算所需的结构,并可在3D显示区域查看其结构的3D视图。搭建好结构后,用户可在LAMMPS计算模块,根据计算需要,在简洁友好的界面中设置参数 生成计算所需的输入文件 ,之后连接装有LAMMPS的 本地电脑或远程服务器 进行相关计算,在计算过程中可实时监测任务的计算状态,计算完成后可对LAMMPS的计算结果进行可视化分析。
目前用户可通过Device Studio生成LAMMPS以下计算输入文件的生成: 结构弛豫、热力学性质(热膨胀系数、体积热容、等压热容)、输运性质(均方位移、速度自相关函数、热导率)、力学性质(杨氏模量、剪切模量)、淬火、退火模拟 ;结合OVITO软件可对LAMMPS计算结果进行结构特征数据分析。
以 对金属铝沿X轴方向以一定的恒定应变速率拉伸的形变模拟 为例来详细描述 LAMMPS 在Device Studio中的应用。

10.2.2. LAMMPS创建项目¶
双击 Device Studio 图标快捷方式启动软件,根据界面提示选择创建一个新的项目( Create a new Project )或打开一个已经存在的项目( Open an existing Project )的按钮,选中之后点击界面中的 OK 按钮即可。若选择创建一个新的项目,用户可根据需要给该项目命名,如本项目命名为 LAMMPS ,或采用软件默认项目名。
10.2.3. LAMMPS导入结构¶
在Device Studio的图形界面中点击 File → Import → Import Local,
则弹出导入LAMMPS结构文件的界面,根据界面提示找到 Al_Rede.hzw 结构文件的位置,选中 Al_Rede.hzw 结构
文件,点击 打开 按钮则导入 Al_Rede.hzw 结构后的Device Studio界面如 图 10.19 所示。
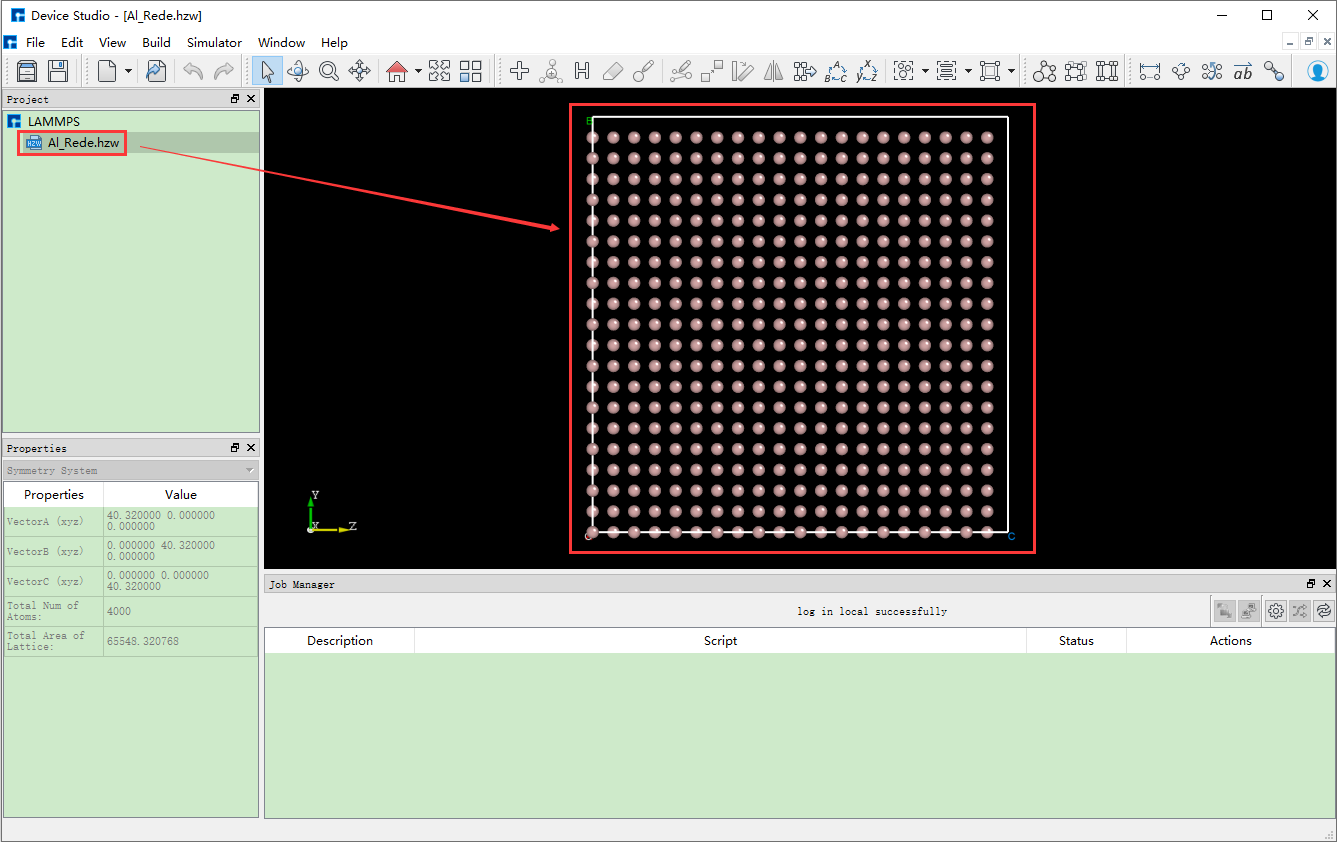
图 10.19 导入 Al_Rede.hzw 结构后的Device Studio图形界面¶
10.2.4. LAMMPS输入文件的生成¶
在如 图 10.19 所示的界面中选中 Simulator → LAMMPS → Calculation, 弹出LAMMPS的参数设置界面LAMMPS Calculation如 图 10.20 所示。LAMMPS的参数设置界面主要分为 Task 、 Force field 、 Setting 和 Run simulation 四个模块,用户可根据 计算需要,依次点击四个模块进行参数设置,之后点击 Generate files 即可生成对应计算的输入文件。
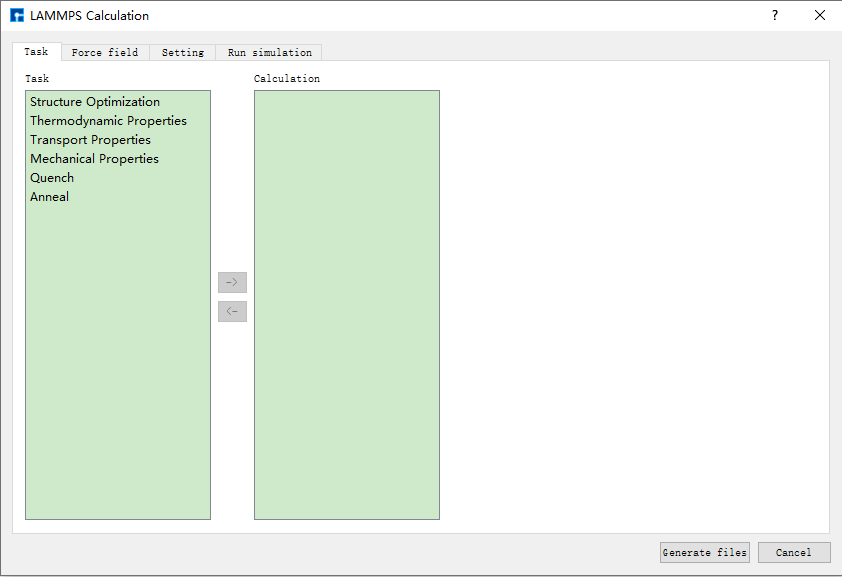
图 10.20 LAMMPS的参数设置界面LAMMPS Calculation¶
以生成 对金属铝沿X轴方向以一定的恒定应变速率拉伸的形变模拟 的输入文件为例,在如 图 10.20 所示的LAMMPS参数设置界
面中,根据计算需要分别选中 Task 、 Force field 、 Setting 和 Run simulation ,设置参数分别如 图 10.21 、 图 10.22 (b)、 图 10.23 (c)和 图 10.24 (d)所示,之后点击界面中的 Generate files 即可生成输入文件 Al_Rede.in、Al_Rede.data 。
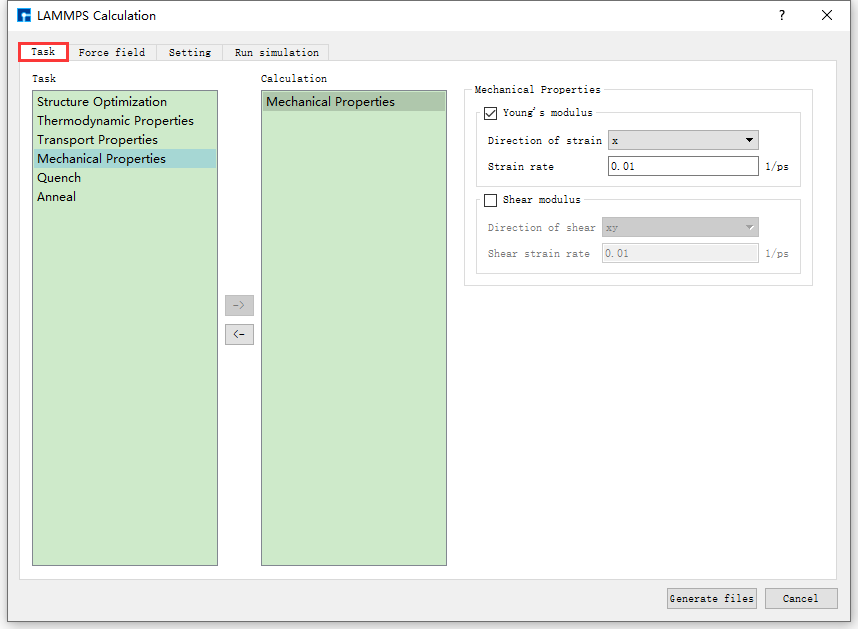
图 10.21 Task参数设置界面¶
Task说明
结构弛豫(Structure Optimization)。
热力学性质(Thermodynamic Properties),其中热力学性质任务包括热膨胀系数(Thermal expansion)、体积热容(Volumetric heat capacity)、等压热容(Isobaric heat capacity)。
输运性质(Transport Properties),其中输运性质任务包括均方位移(MSD)、速度自相关函数(VACF)、热导率(Thermal conductivity)。
力学性质(Mechanical Properties),其中力学性质包括杨氏模量(Young’s modulus)、剪切模量(Shear modulus)。
淬火模拟(Quench)。
退火模拟(Anneal)。
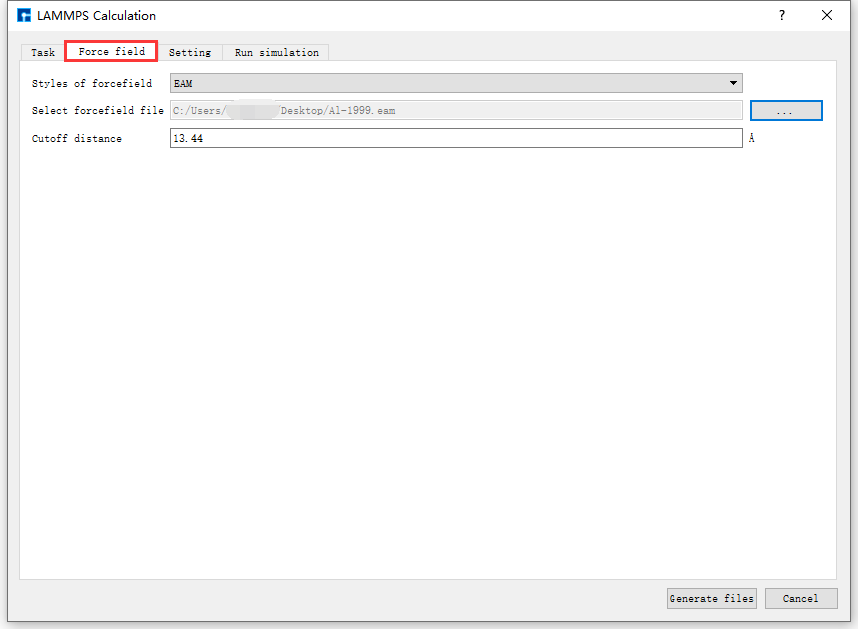
图 10.22 Force field参数设置界面¶
Force field说明
在 Force field 模块,目前用户可选择EAM和MEAM两种力场类型,用户需提前自行准备力场文件,并在 Select forcefield file 部分选定所需力场文件。
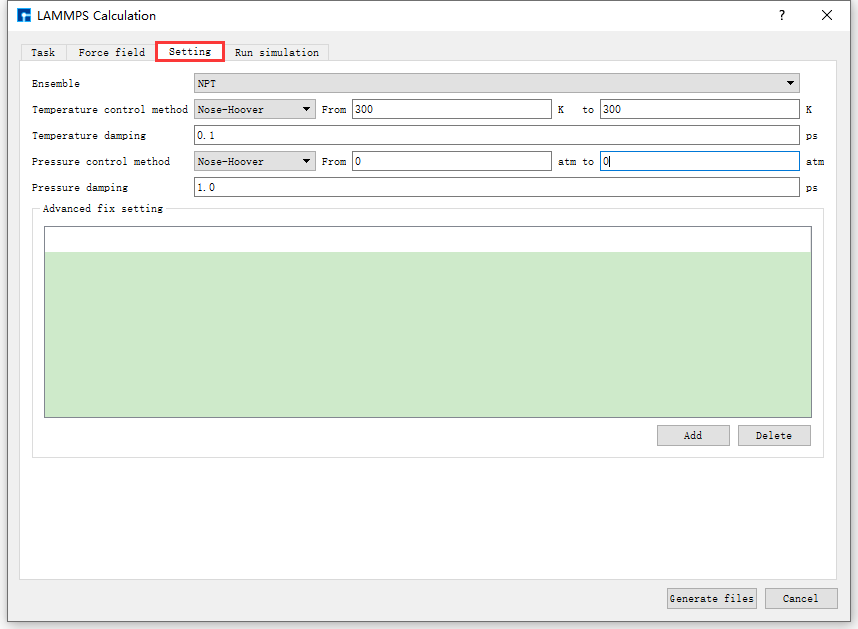
图 10.23 Setting参数设置界面¶
图 10.24 Run simulation参数设置界面¶
生成 对金属铝沿X轴方向以一定的恒定应变速率拉伸的形变模拟 的输入文件 Al_Rede.in、Al_Rede.data 的Device Studio界面如 图 10.25 所示。
图 10.25 生成 对金属铝沿X轴方向以一定的恒定应变速率拉伸的形变模拟 的输入文件的Device Studio界面¶
10.2.5. LAMMPS计算¶
在做 对金属铝沿X轴方向以一定的恒定应变速率拉伸的形变模拟 之前,需连接装有LAMMPS的本地电脑或服务器,具体连接过程这里
不做详细说明,用户可参考 Nanodcal连接服务器 节内容。这里以在本地电脑上计算为例,连接好装有LAMMPS的本地电脑后,在做
计算之前,用户可根据需要打开输入文件并查看文件中的参数设置是否合理,若不合理,则可选择直接在文件中编辑或重新生成,最后再进
行LAMMPS计算。如打开 Al_Rede.in 文件,在Device Studio的Project Explorer区域选中 Al_Rede.in → 右击 →
Open with 即可查看到 Al_Rede.in 文件如 图 10.26 所示。对于其他输入文件,用户可根据需要选择是否打开查看,
这里不做详细说明。
图 10.26 Al_Rede.in 文件¶
在如 图 10.25 所示界面中,在Device Studio的的Project Explorer区域选中 Al_Rede.in → 右击 → Run ,弹出Run
界面,在Run界面中点击 Run 按钮则可做LAMMPS计算。用户可在Job Manager区域中观察LAMMPS的计算状态,当LAMMPS计算
任务处于排队中、计算中和计算完成时,Status 分别为Queued、Running、Finished。计算完成后,选中 Al_Rede.in → 右击 → Open Containing Folder 可看到LAMMPS计算完成的结果文件 Young-Stress.dat 和 Al_Rede-tension-300K.dump ,并找到其在本地电脑中存储位置。

10.3. QUANTUM ESPRESSO 实例¶
QUANTUM ESPRESSO 是一款功能很强大的第一性原理计算开源软件,ESPRESSO是opEn-Source Package的首字母缩写,基于密度泛函理论、平面波及赝势,可用于纳米尺度下电子结构计算。 QUANTUM ESPRESSO 的核心平面波DFT模块为PWscf(平面波自洽场),PWscf(平面波自洽场)使用平面波基集和伪势能在密度泛函理论和密度泛函微扰理论范围内进行电子结构计算的程序。 目前,PWscf(平面波自洽场)模块已经集成在Device Studio中。
QUANTUM ESPRESSO软件官网:详见 http://www.quantum-espresso.org/
目前用户可通过Device Studio进行 QUANTUM ESPRESSO 以下性质的计算: 结构驰豫、自洽、能带、态密度、DFT+U、共线自旋、非共线自旋、自旋轨道耦合。
以 Si晶体结构的自洽、能带及态密度计算 为例来详细描述 QUANTUM ESPRESSO 在Device Studio中的应用。
10.3.1. QUANTUM ESPRESSO计算流程¶
QUANTUM ESPRESSO在Device Studio中的计算流程如 图 10.28 所示。
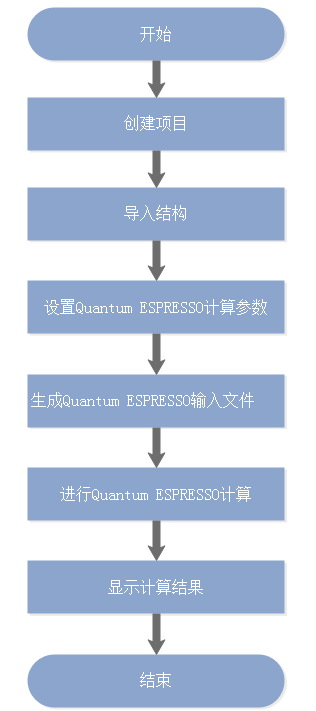
图 10.28 QUANTUM ESPRESSO计算流程¶
10.3.2. QUANTUM ESPRESSO创建项目¶
双击Device Studio图标快捷方式,登录并启动Device Studio,在创建或打开项目界面中(启动软件后选择创建或打开项目的图形界面),根据界面提示选择创建一个新的项目( Create a new Project )或打开一个已经存在的项目( Open an existing Project )的按钮,选中之后点击界面中的 OK 按钮即可。若选择创建一个新的项目,用户可根据需要给该项目命名,如本项目命名为 QUANTUM ESPRESSO,或采用软件默认项目名。
10.3.3. QUANTUM ESPRESSO导入结构¶
在Device Studio中导入Si晶体结构后的图形界面如 图 10.29 。在Device Studio中导入Si晶体结构的具体操作这里不做详细说明,用户可参照 导入结构 节内容。
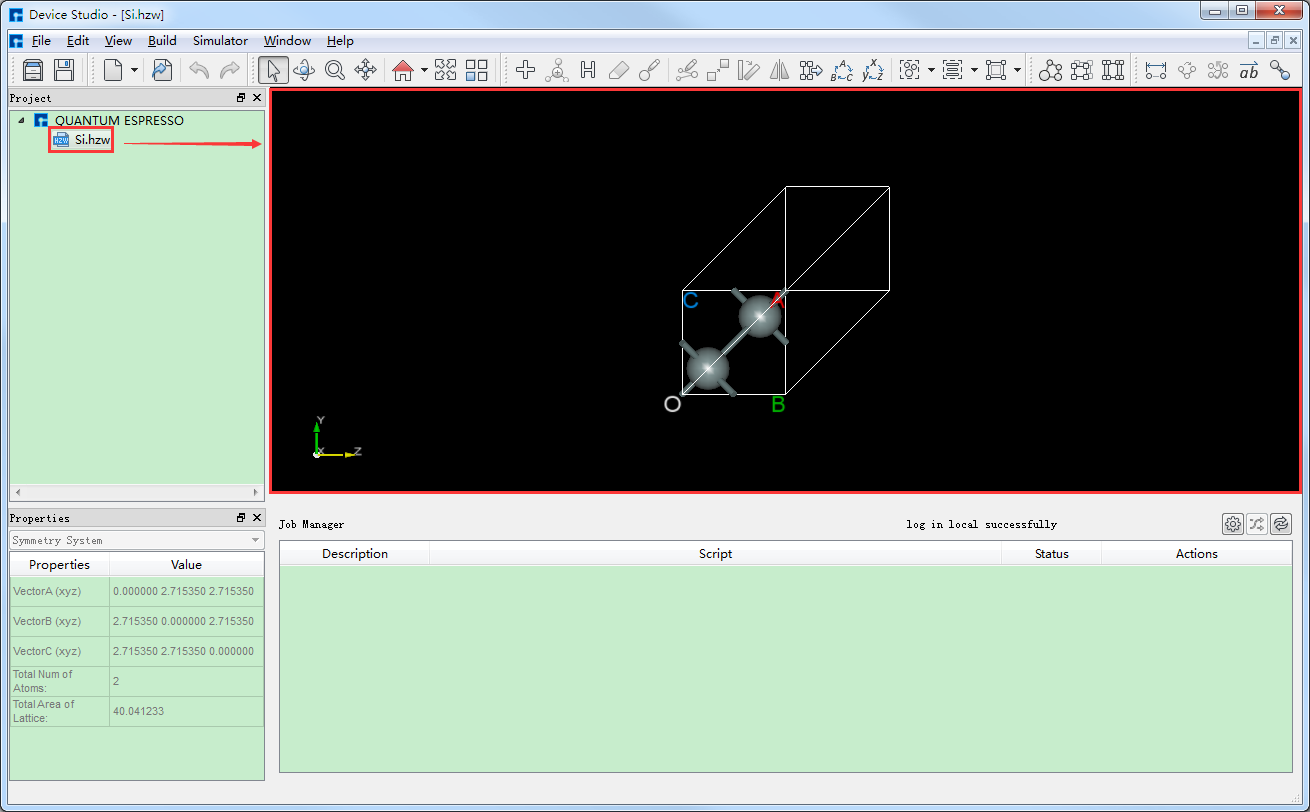
图 10.29 导入Si晶体结构后的Device Studio图形界面¶
10.3.4. QUANTUM ESPRESSO参数设置¶
在如 图 10.29 所示界面中选中 Simulator → QUANTUM ESPRESSO → QUANTUM ESPRESSO,弹出界面如 图 10.30 所示,用户可根据所计算的结构及计算需要在该界面中点击不同的按钮合理设置参数,之后点击 Generate files 即可生成对应计算的输入文件。
如生成Si晶体结构的自洽、能带及态密度计算的输入文件,根据计算需要设置参数,分别选中 Basic settings 、Bandstructure、DensityOfStates,设置参数分别如 图 10.31 、 图 10.32 、 图 10.33 所示,设置好参数后点击 Generate files 即可生成Si晶体结构自洽、能带及态密度计算的输入文件 scf.in、band.in、nscf.in、dos.in。
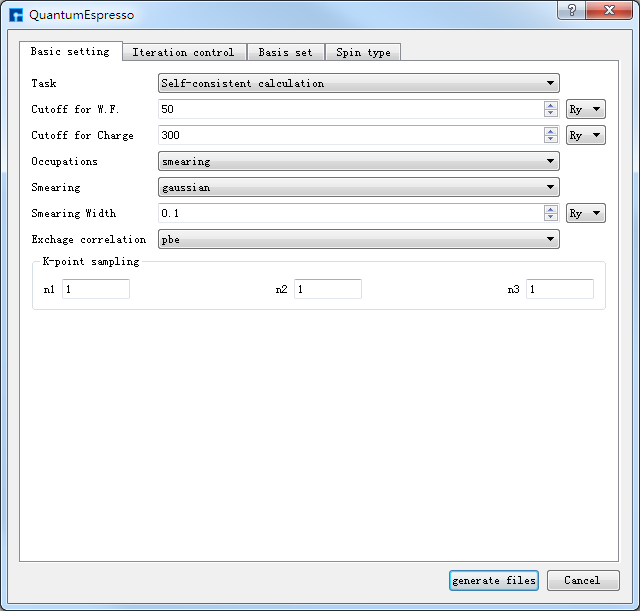
图 10.30 QUANTUM ESPRESSO界面¶ |
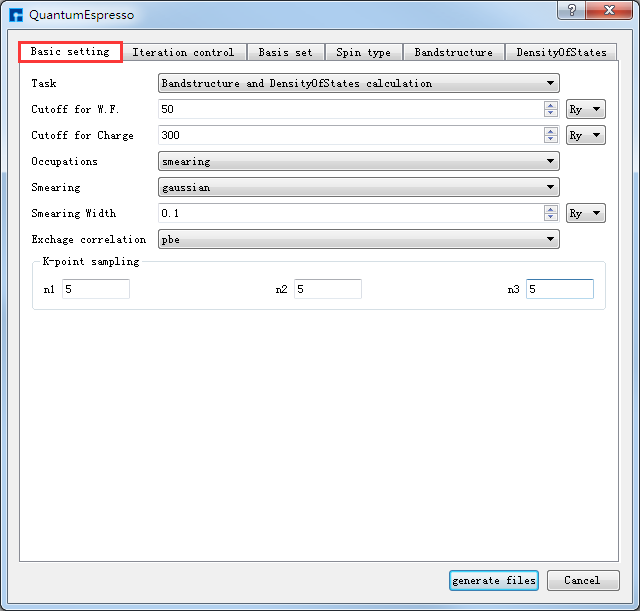
图 10.31 Basic settings参数设置界面¶ |
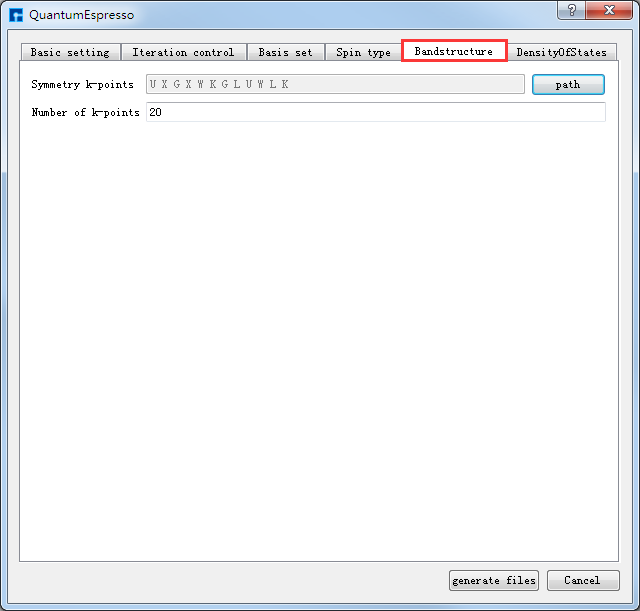
图 10.32 Bandstructure参数设置界面¶ |
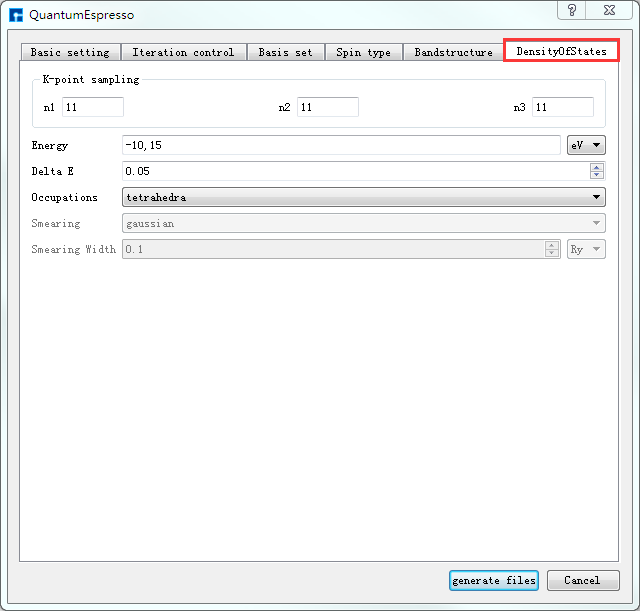
图 10.33 DensityOfStates参数设置界面¶ |
10.3.5. QUANTUM ESPRESSO输入文件的生成¶
生成Si晶体结构的自洽、能带和态密度计算的输入文件 scf.in、band.in、nscf.in、dos.in 的Device Studio界面如 图 10.34 所示。其中,scf.in 为自洽计算输入文件; band.in 为能带计算输入文件;由于态密度计算之前需要先进行一个非自洽计算,因此 nscf.in 和 dos.in 是态密度计算所需的输入文件,计算顺序为先 nscf.in 后 dos.in 。
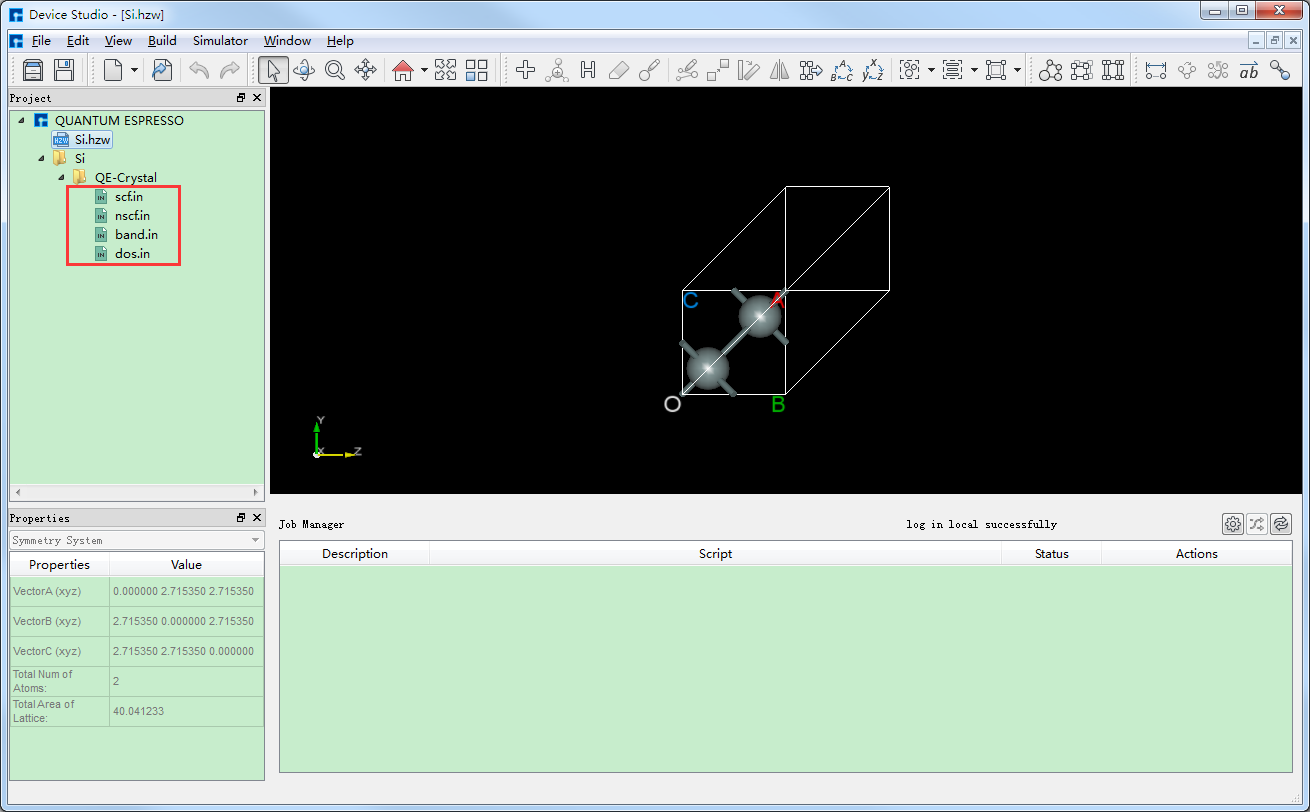
图 10.34 生成Si晶体结构自洽、能带、态密度计算输入文件的Device Studio图形界面¶
在如 图 10.34 所示界面中,选中 scf.in → 右击 → Open with 即可查看到Si晶体结构自洽计算输入文件如下所示。对于其他输入文件,用户可根据计算需要选择是否打开查看,这里不做详细说明。
&CONTROL
calculation='scf',
outdir='results',
prefix='Si',
pseudo_dir='../',
verbosity='high',
/
&SYSTEM
ibrav=0,
nat=2,
ntyp=1,
ecutwfc=50,
ecutrho=300,
input_dft='pbe',
occupations='smearing',
smearing='gaussian',
degauss=0.1,
/
&ELECTRONS
diagonalization='david',
conv_thr=1.D-6,
mixing_mode='plain',
mixing_beta=0.7D0,
electron_maxstep=200
/
ATOMIC_SPECIES
Si 28.0855 Si.pbe-n-kjpaw_psl.1.0.0.UPF
CELL_PARAMETERS {angstrom}
0 2.71535 2.71535
2.71535 0 2.71535
2.71535 2.71535 0
ATOMIC_POSITIONS {angstrom}
Si 0.678837 0.678837 0.678837
Si 2.03651 2.03651 2.03651
K_POINTS {automatic}
5 5 5 0 0 0
10.3.6. QUANTUM ESPRESSO计算¶
在如 图 10.34 所示界面中,在Device Studio的Project Explorer区域选中 scf.in → 右击 → Run,弹出Run界面如 图 10.35 所示,点击界面中的 Run 按钮则可进行Si晶体结构的自洽计算。以此类推,分别选中 band.in、nscf.in、dos.in 文件重复上述步骤进行Si的能带和态密度计算。
在计算过程中,用户可在Job Manager区域观察自洽、能带和态密度计算的计算状态,当计算任务处于排队中、计算中和计算完成时,Status 分别为Queued、Running、Finished,计算完成后Device Studio图形界面如 图 10.36 所示。其中,scf.out、bs.out 和 nscf.out 分别为Si晶体结构自洽、能带和非自洽计算的日志文件;dos.out 和 Si.dos 分别为Si态密度计算的日志文件和数据文件。
图 10.35 Run界面¶
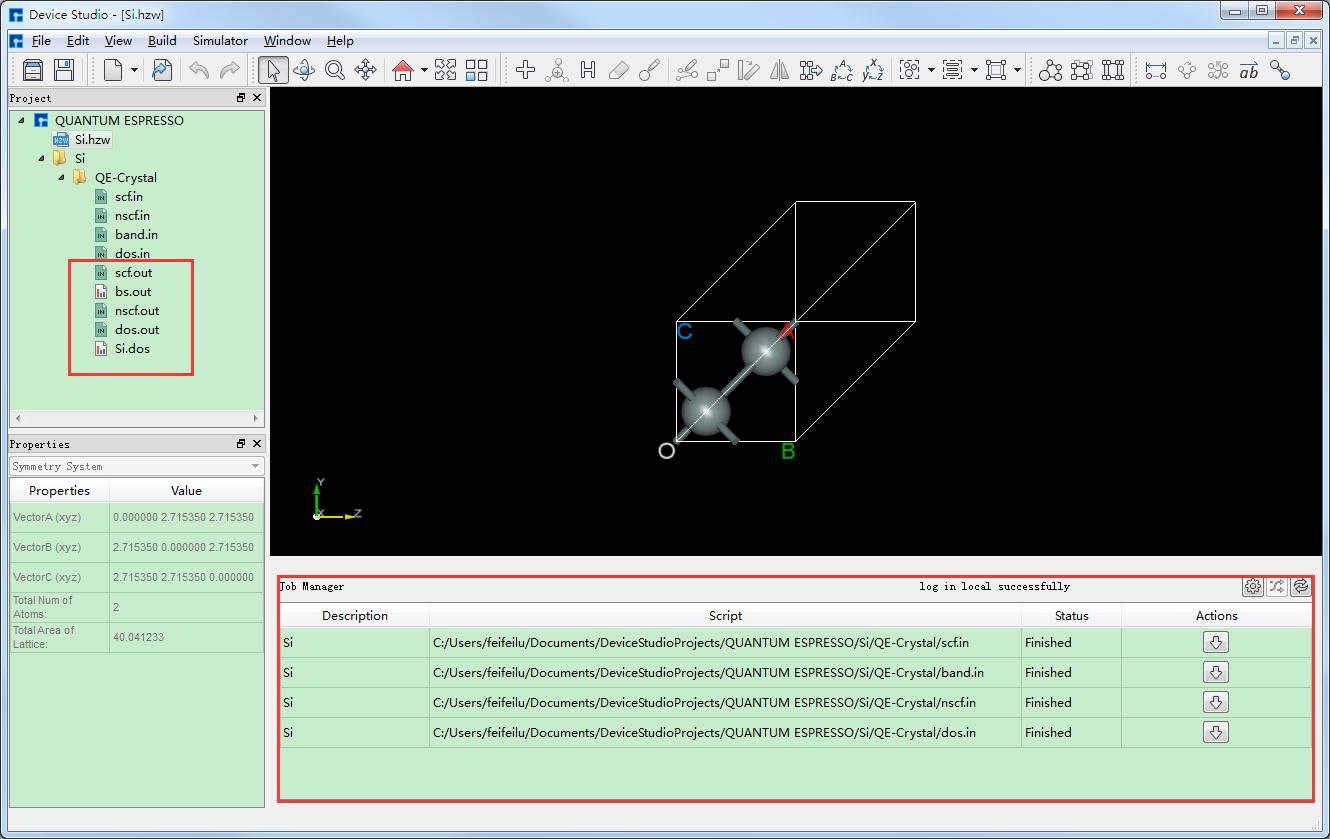
图 10.36 Si晶体结构自洽、能带和态密度计算完成的Device Studio图形界面¶
10.3.7. QUANTUM ESPRESSO计算结果的可视化分析¶
对于Si的能带和态密度计算结果 bs.out 和 Si.dos,Device Studio可将其进行显示供用户进行相关分析,即计算结果的可视化分析。如 bs.out,在如 图 10.36 所示界面的Project Explorer区域选中 bs.out → 右击 → Show View,则弹出Si的能带可视化分析界面如 图 10.37 所示,用户可通过滚动鼠标中键将可视化分析结果放大或缩小。选中如 图 10.37 所示界面中 Export 快捷图标,弹出导出可视化分析结果的图形界面,用户可根据需要选择图片的保存路径和保存格式,并给所保存的图片命名。
在Device Studio的Project Explorer区域选中 Si.dos → 右击 → Show View,则弹出Si的态密度可视化分析界面如 图 10.38 所示。
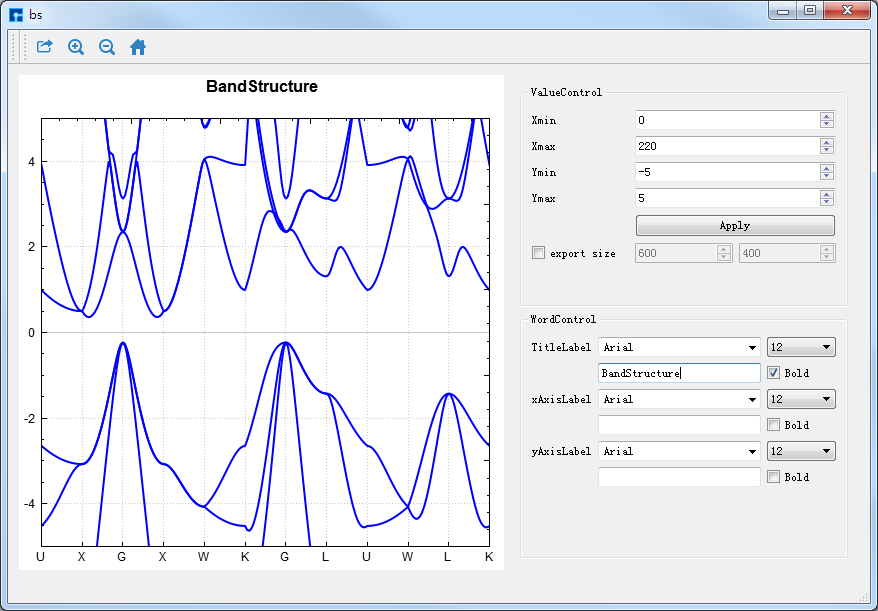
图 10.37 Si晶体能带( bs.out )的可视化分析界面¶
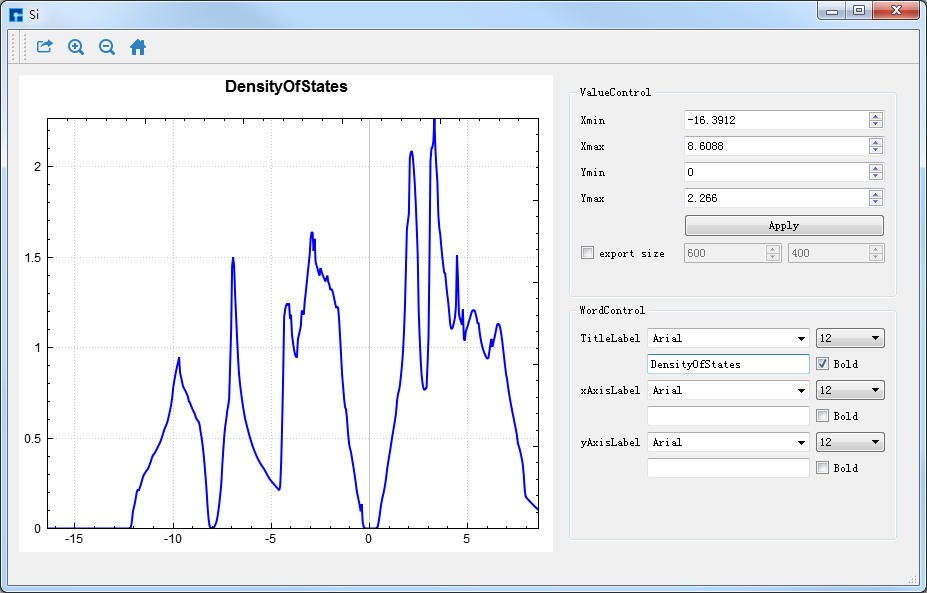
图 10.38 Si晶体态密度( Si.dos )的可视化分析界面¶
10.4. RESCU 实例¶
RESCU 是一款仅仅用小型计算机就能研究超大体系的KS-DFT计算软件。RESCU 是Real Space Electronic Structure CalcUlator(实空间电子结构计算程序)的缩写,它的核心是一种全新、极其强大、并行效率超高的KS-DFT自洽计算方法。
RESCU 可以使用各类计算机资源,从笔记本电脑、桌面单机、到16核、64核、256核、到更大的超算、包括用GPU加速等等,来计算包含一千、数千、上万、乃至更大体系的电子结构性质。RESCU是解决超大体系KS-DFT问题的新方法,正在被应用于金属、半导体、绝缘体、液体、DNA、1维、2维、3维、表面、分子、磁性、非磁性、杂质、固体等不同系统的KS-DFT计算。
这里以 晶体Ni的自洽、能带、态密度计算 为例详细描述 RESCU 软件在Device Studio中的应用。包含创建项目,从建模到计算,到数据的可视化分析整个流程。实际上, RESCU 的功能不仅于此,详情可参照 RESCU 应用教程或点击对应的紫色或蓝色字体软件名称。Device Studio可以生成 RESCU 很多功能计算的输入文件,用户可根据计算需要选择生成。

10.4.2. RESCU创建项目¶
双击Device Studio图标快捷方式,登录并启动Device Studio,在创建或打开项目界面中(启动软件后选择创建或打开项目的图形界面),根据界面提示选择创建一个新的项目( Create a new Project )或打开一个已经存在的项目( Open an existing Project )的按钮,选中之后点击界面中的 OK 按钮即可。若选择创建一个新的项目,用户可根据需要给该项目命名,如本项目命名为 RESCU,或采用软件默认项目名。
10.4.3. RESCU导入结构¶
在Device Studio中导入晶体Ni后的界面如 图 10.40 。在Device Studio中导入晶体Ni的具体操作这里不做详细说明,用户可参照 导入结构 节内容。
图 10.40 导入晶体Ni后的Device Studio图形界面¶
10.4.4. RESCU输入文件的生成¶
10.4.4.1. RESCU生成自洽计算输入文件¶
在如 图 10.40 所示界面中选中 Simulator → RESCU → SCF Calculation,弹出界面如 图 10.41 所示,用户可根据所计算的结构及计算需要分别选中 Basic settings 、Iteration control、Basis set、Spin type 这四个按钮合理设置参数,之后点击 Generate files 即可生成自洽计算输入文件。
如生成晶体Ni自洽计算输入文件,根据计算需要设置参数,分别选中 Basic settings 、Iteration control、Basis set、Spin type 设置参数分别如 图 10.41 、 图 10.42 、 图 10.43 和 图 10.44 所示,设置好参数后点击 Generate files 即可生成晶体Ni自洽计算输入文件 scf.input 及原子坐标文件 Atom.xyz。
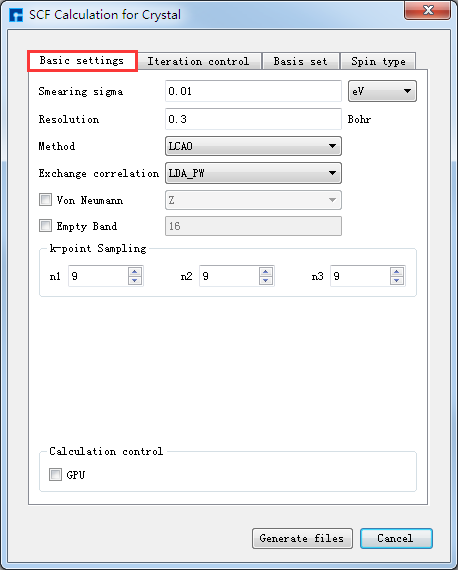
图 10.41 Basic settings参数设置界面¶ |
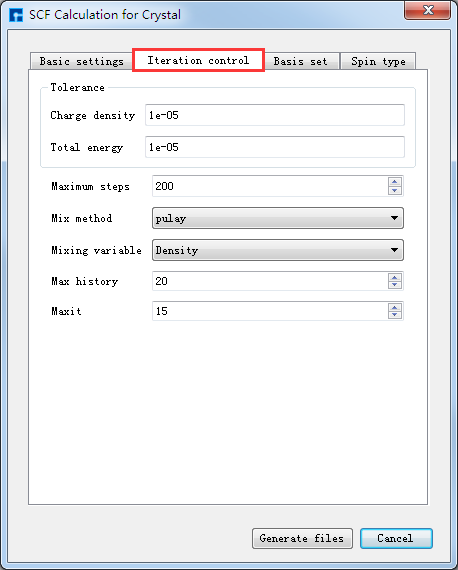
图 10.42 Iteration control参数设置界面¶ |
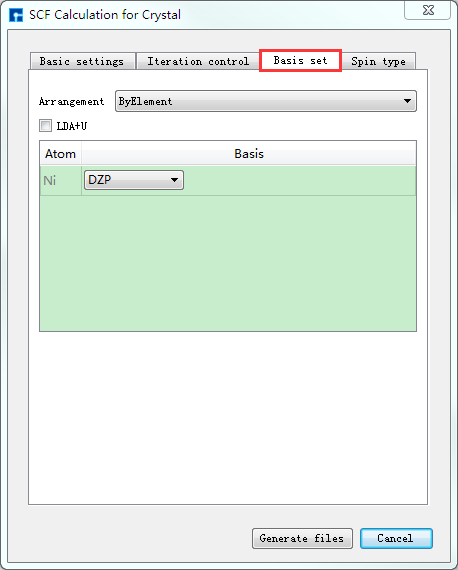
图 10.43 Basis set参数设置界面¶ |
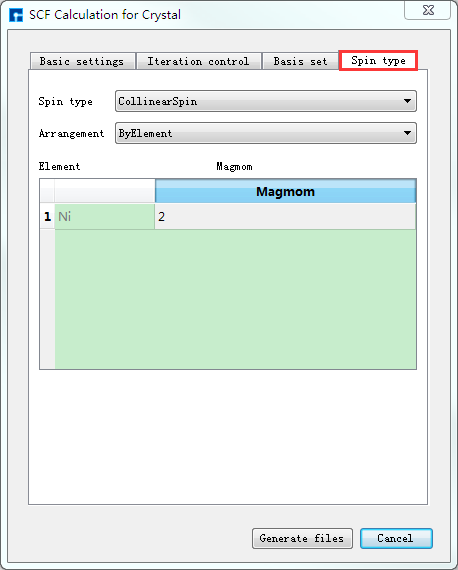
图 10.44 Spin type参数设置界面¶ |
10.4.4.2. RESCU生成能带计算输入文件¶
在如 图 10.40 所示界面(即:Device Studio图形界面)中选中 Simulator → RESCU → Analysis,弹出Analysis界面,在界面中双击 BandStructure,点击 path 按钮弹出Brillouin Zone Path参数设置界面,设置高对称点为 W-L-G-X-W-K ,并点击 Apply 如 图 10.45 所示。能带计算的参数设置如 图 10.46 所示,设置好参数后点击 Generate files 即可生成晶体Ni能带计算输入文件 BandStructure.input。
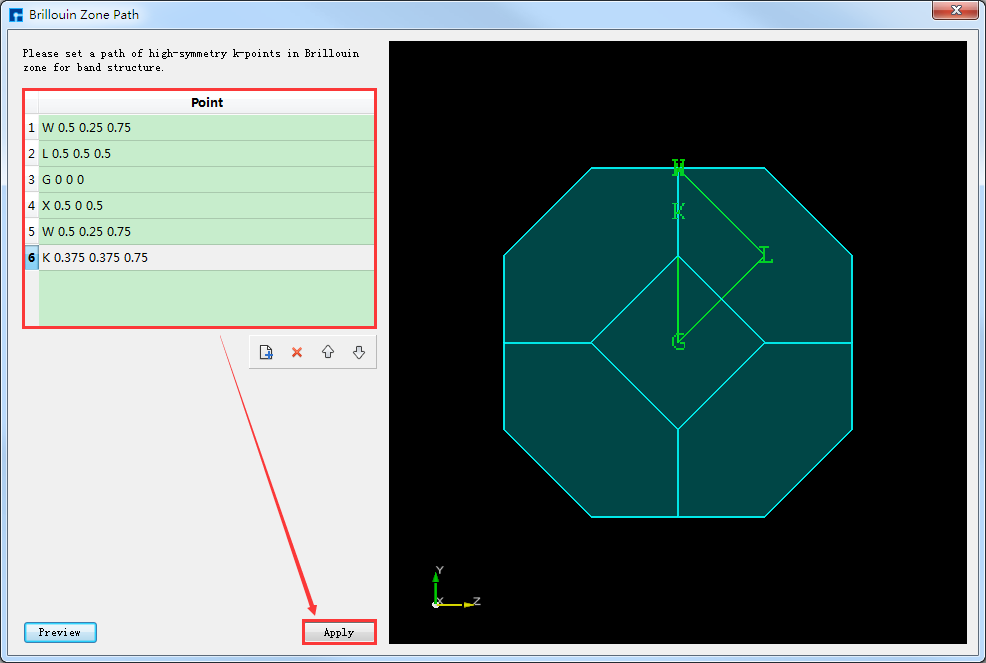
图 10.45 Brillouin Zone Path参数设置界面¶
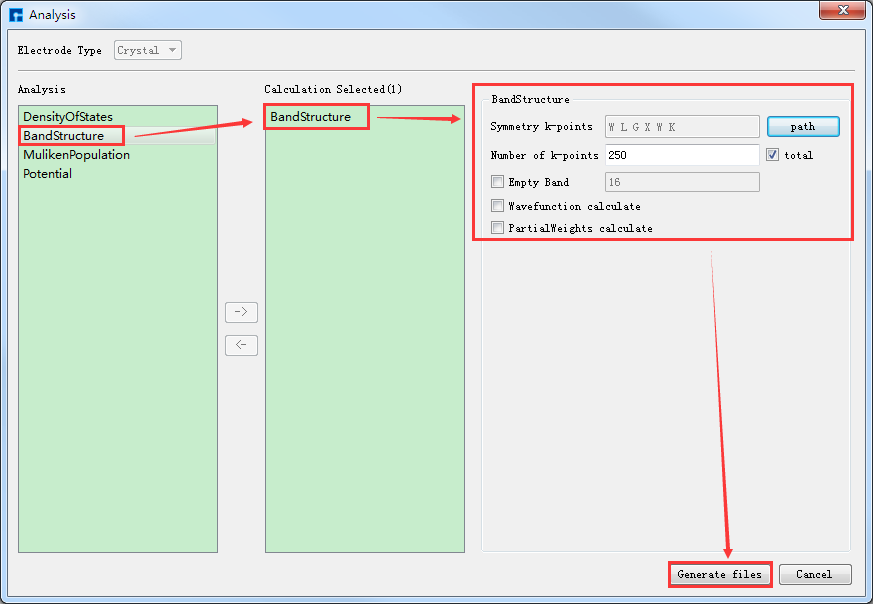
图 10.46 晶体Ni能带计算参数设置界面¶
10.4.5. RESCU计算¶
10.4.5.1. RESCU自洽计算¶
在做自洽计算之前,用户可根据计算需要打开 scf.input 文件并查看文件中的参数设置是否合理,若不合理,则可选择直接在文件中进行编辑或重新生成,最后再进行自洽计算。选中 scf.input → 右击 → Open with 即可查看到晶体Ni自洽计算输入文件如下所示:
info.calculationType = 'self-consistent'
info.savepath = './results/Ni_scf'
%symmetry.spacesymmetry = 0
domain.latvec = [[0 1.762 1.762]; [1.762 0 1.762]; [1.762 1.762 0]]
%eigensolver.emptyBand = 16
eigensolver.maxit = 15
eigensolver.algo = 'cfsi'
LCAO.status = 1
smearing.sigma = 0.01
units.energy = 'eV'
kpoint.gridn = [9,9,9]
domain.lowres = 0.3
%domain.highres = 0.3
functional.libxc = 1
functional.list = {'XC_LDA_X','XC_LDA_C_PW'}
option.maxSCFiteration = 200
mixing.type = 'density'
mixing.method = 'pulay'
mixing.tol = [1e-05,1e-05]
mixing.maxhistory = 20
spin.type = 'collinear'
atom.magmom = 'Atom.xyz'
element(1).species = 'Ni'
element(1).path = '../Ni_DZP.mat'
atom.xyz = 'Atom.xyz'
units.length = 'Angstrom'
gpu.status = 0
在Device Studio的Project Explorer区域选中 scf.input → 右击 → Run,弹出Run界面如 图 10.48 Run界面 所示,点击界面中的 Run 按钮则可进行晶体Ni的自洽计算。
在计算过程中,用户可在Job Manager区域观察晶体Ni自洽计算的计算状态,当计算任务处于排队中、计算中和计算完成时,Status 分别为Queued、Running、Finished。计算完成后可看到Device Studio的Project Explorer区域看到日志文件 resculog.out。选中 resculog.out → 右击 → Open Containing Folder 则找到 resculog.out 文件在本地电脑中的存储位置,其中results文件夹则含有晶体Ni自洽计算完成后的结果文件 Ni_scf.h5、 Ni_scf.mat 和 Density.txt 。
备注
对于RESCU计算,选中其在Device Studio的Project Explorer区域任何一个输入文件 → 右击 → Open Containing Folder 均可找到该输入文件在本地电脑中的存储位置,若计算完成,可在与输入文件同一目录下找到results文件,results文件中存储着计算结果。
图 10.48 Run界面¶
10.4.5.2. RESCU能带计算¶
能带计算需调用自洽计算结果,故需先做自洽计算,再做能带计算。本算例已经做过自洽计算,则不需要再做自洽计算,直接调用自洽计算结果做能带计算即可。选中 Bandstructure.input → 右击 → Open with 即可查看到晶体Ni能带计算输入文件如下所示:
info.calculationType = 'band-structure'
info.savepath = './results/Ni_bs'
rho.in = './results/Ni_scf'
LCAO.status = 1
domain.bravaisLattice= 'FCC'
kpoint.gridn = 250
kpoint.sympoints = {'W','L','G','X','W','K'}
%kpoint.sympoints = {[0.5 0.25 0.75],[0.5 0.5 0.5],[0 0 0],[0.5 0 0.5],[0.5 0.25 0.75],[0.375 0.375 0.75]}
%eigensolver.emptyBand = 16
在Device Studio的Project Explorer区域选中 Bandstructure.input → 右击 → Run,弹出Run界面如 图 10.48 Run界面 所示,点击界面中的 Run 按钮则可进行晶体Ni的能带计算。计算完成后则可在results文件夹中看到晶体Ni能带计算完成后的结果文件 Ni_bs.h5、 Ni_bs.mat 和 BandStructure.txt 。
10.4.5.3. RESCU态密度计算¶
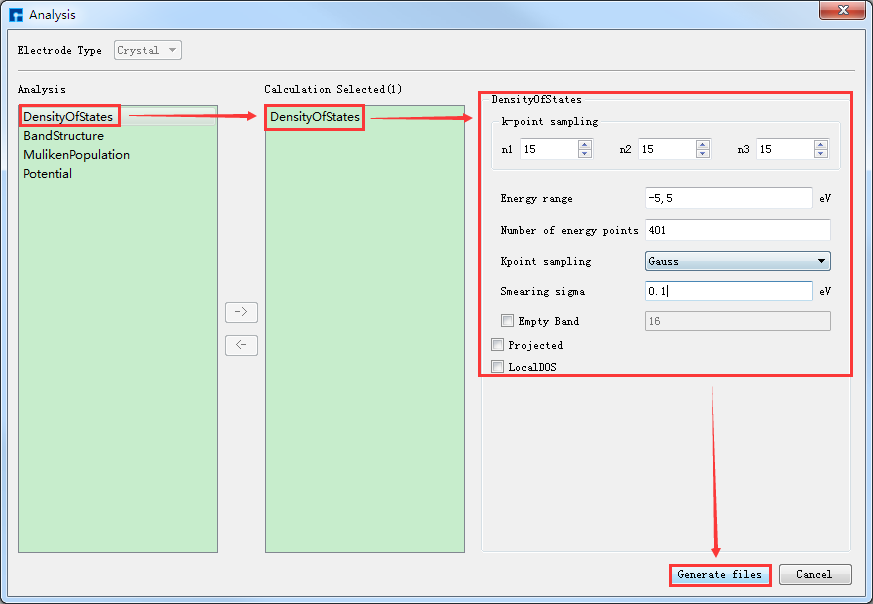
态密度计算需调用自洽计算结果,故需先做自洽计算,再做态密度计算。本算例已经做过自洽计算,则不需要再做自洽计算,直接调用自洽计算结果做态密度计算即可。选中 DensityOfStates.input → 右击 → Open with 即可查看到晶体Ni态密度计算输入文件如下所示:
info.calculationType = 'dos'
info.savepath = './results/Ni_dos'
rho.in = './results/Ni_scf'
LCAO.status = 1
smearing.sigma = 0.1
units.energy = 'eV'
kpoint.gridn = [ 15,15,15 ]
kpoint.sampling = 'gauss'
dos.range = [-5 , 5]
dos.resolution = 0.025
%eigensolver.emptyBand = 16
在Device Studio的Project Explorer区域选中 DensityOfStates.input → 右击 → Run,弹出Run界面,点击界面中的 Run 按钮则可进行晶体Ni的态密度计算。计算完成后则可在results文件夹中看到晶体Ni态密度计算完成后的结果文件 Ni_dos.h5、 Ni_dos.mat 和 DensityOfStates.txt 。
10.4.6. RESCU计算结果的可视化分析¶
选中 resculog.out → 右击 → Open Containing Folder 则找到 resculog.out 文件在本地电脑中的存储位置,在同一目录下找到计算结果文件夹results,该文件夹下含有晶体Ni的自洽、能带和态密度计算的结果文件。
在如 图 10.40 所示界面(即:Device Studio图形界面)中选中 Simulator → RESCU → Analysis Plot,弹出导入计算结果文件的图形界面如 图 10.49 所示,在该界面中选中能带计算结果文件 Bandstructure.txt ,点击 打开 按钮即可弹出能带的可视化分析界面如 图 10.50 所示。用户可根据需要调节坐标轴的取值范围,通过点击界面中的 Spin Type 的下拉按钮选择只显示Spin Up或Spin Down,或Spin Up和Spin Down都显示。
用户可通过滚动鼠标中键将可视化分析结果放大或缩小;可根据需要编辑坐标轴标注和标题,选择字体的大小和类型。选中如 图 10.50 所示界面中 Export 快捷图标,弹出导出可视化分析结果的图形界面,用户可根据需要选择图片的保存路径和保存格式,并给所保存的图片命名。
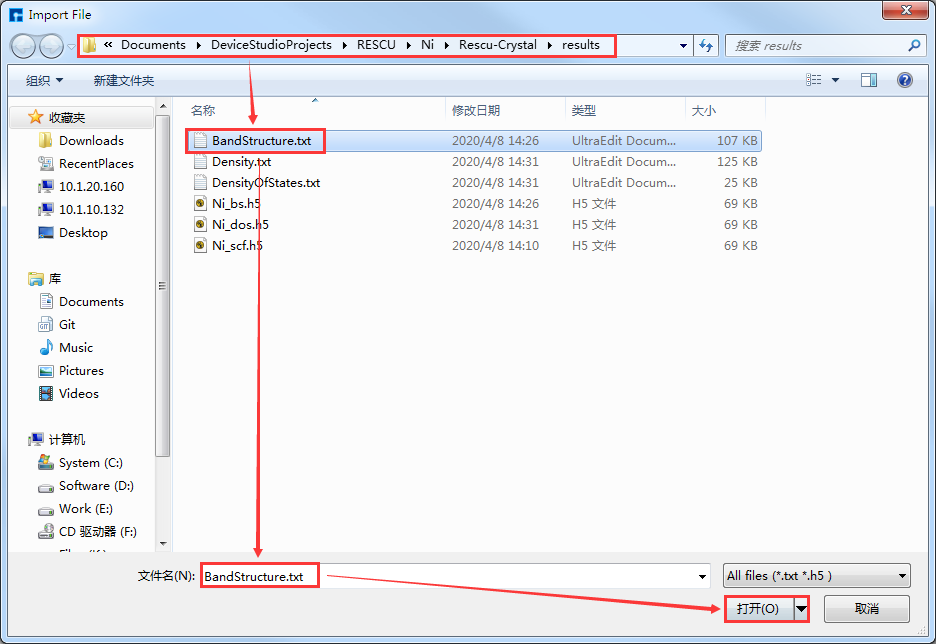
图 10.49 RESCU导入计算结果文件的图形界面¶
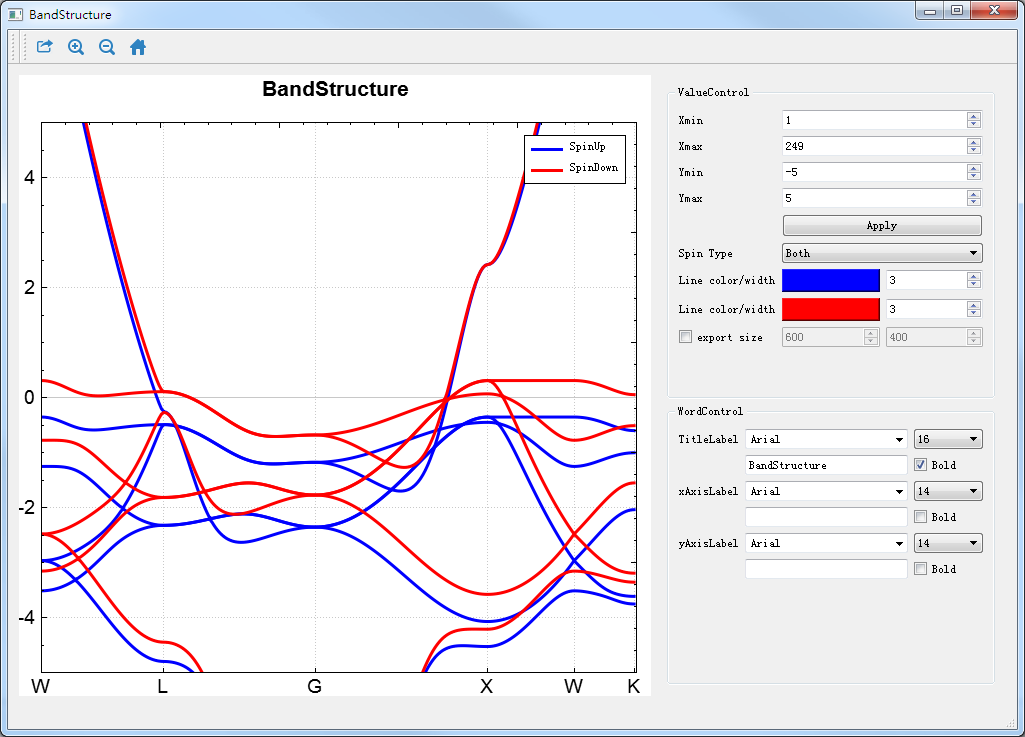
图 10.50 RESCU能带的可视化分析界面¶
在如 图 10.40 所示界面(即:Device Studio图形界面)中选中 Simulator → RESCU → Analysis Plot,弹出导入计算结果文件的图形界面如 图 10.49 所示,在该界面中选中能带计算结果文件 DensityOfStates.txt ,点击 打开 按钮即可弹出态密度的可视化分析界面如 图 10.51 所示。
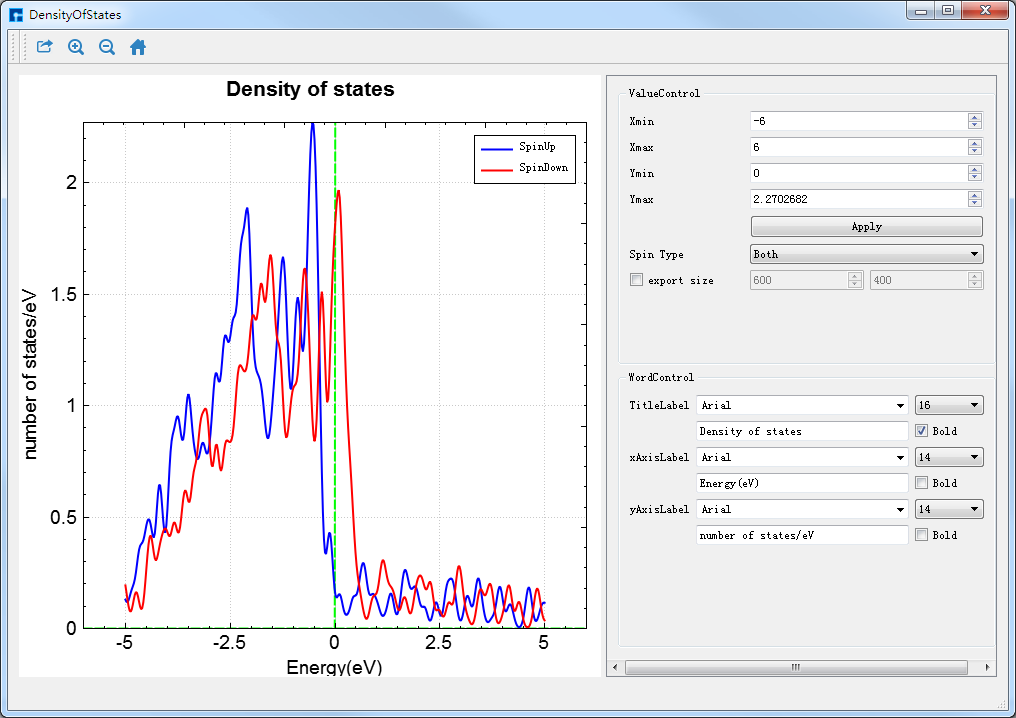
图 10.51 RESCU态密度的可视化分析界面¶
10.5. Nanoskim 实例¶
Nanoskim 即 Nanostructure Slater-Koster Integrals Manifesting package ,是一款基于Slater-Koster紧束缚势场的量子输运仿真软件,可以从原子层级来构建纳米器件。在非平衡条件下,通过自洽求解非平衡格林函数(Non-equilibrium Green’s function, 简称:NEGF)和,得到纳米器件的电子密度和静电势,从而可以充分考虑量子力学效应(如源漏隧穿效应、量子受限效应),以及具体的原子细节(如截面形状、杂质位置)等。
Nanoskim软件官网:详见 https://cloud.hzwtech.com/web/product-service?id=4
以 环栅型的硅纳米线晶体管结构的电流电压特性 为例详细描述 Nanoskim 软件在Device Studio中的应用。

10.5.2. Nanoskim创建项目¶
双击Device Studio图标快捷方式,登录并启动Device Studio,在创建或打开项目界面中(启动软件后选择创建或打开项目的图形界面),根据界面提示选择创建一个新的项目( Create a new Project )或打开一个已经存在的项目( Open an existing Project )的按钮,选中之后点击界面中的 OK 按钮即可。若选择创建一个新的项目,用户可根据需要给该项目命名,如本项目命名为 Nanoskim,或采用软件默认项目名。
10.5.3. Nanoskim导入结构¶
在Device Studio中导入纯硅器件结构( Device_Si.hzw )后的图形界面如 图 10.53 。在Device Studio中导入纯硅器件结构的具体操作这里不做详细说明,用户可参照 导入结构 节内容。
图 10.53 导入纯硅器件结构( Device_Si.hzw )后的Device Studio图形界面¶
10.5.4. Nanoskim输入文件的生成¶
10.5.4.1. Nanoskim生成准备文件¶
在如 图 10.53 所示界面中选中 Simulator → Nanoskim → Preparation,弹出界面如 图 10.54 所示,用户可根据所计算的结构及计算需要分别选中 Hamiltonian、Bandstructure 这2个按钮合理设置参数,之后点击 Generate files 即可生成准备文件。
如生成纯硅器件结构的准备文件,分别选中 Hamiltonian、Bandstructure 设置参数分别如 图 10.54 、 图 10.55 所示,设置好参数后点击 Generate files 即可生成纯硅器件结构的准备文件 Structure.input 、 Hamiltonian.input 、 BandStructure.input。
图 10.54 Hamiltonian参数设置界面¶ |
图 10.55 Bandstructure参数设置界面¶ |
10.5.4.2. Nanoskim生成自洽输入文件¶
在如 图 10.53 所示界面(即:Device Studio图形界面)中选中 Simulator → Nanoskim → SCf Calculation,弹出SCf Calculation界面,在界面中分别选中 Basis settings 、Contour integral、Boundary conditions 设置参数分别如 图 10.56 、 图 10.57 、 图 10.58 所示,设置好参数后点击 Generate files 即可生成纯硅器件结构自洽的输入文件 SCF.input。
备注
本算例生成自洽输入文件均采用的Device Studio的默认参数,实际计算过程中用户通过 Nanoskim 快速使用说明手册 了解各参数的详细意义,从而根据计算需要合理设置参数。若想详细了解 Nanoskim ,点击对应紫色或蓝色字体软件名称,或发送邮件到邮箱 support@hzwtech.com 咨询。
图 10.56 Basis settings参数设置界面¶
图 10.57 Contour integral参数设置界面¶
图 10.58 Boundary conditions参数设置界面¶
10.5.4.3. Nanoskim生成电流、电子透射谱输入文件¶
在如 图 10.53 所示界面(即:Device Studio图形界面)中选中 Simulator → Nanoskim → Analysis,弹出Post Analysis界面,在界面中先双击 Transmission,再双击 IVcurve,并设置参数如 图 10.59 所示,设置好参数后点击 Generate files 即可生成纯硅器件结构电流、电子透射谱输入文件 IVcurve.input 和 Transmission.input。
备注
在Post Analysis界面左侧双击 Transmission,会发现不需要设置相关参数,属于正常现象,Device Studio已自动设置好相关参数。在双击 IVcurve 之前一定要先双击 Transmission。
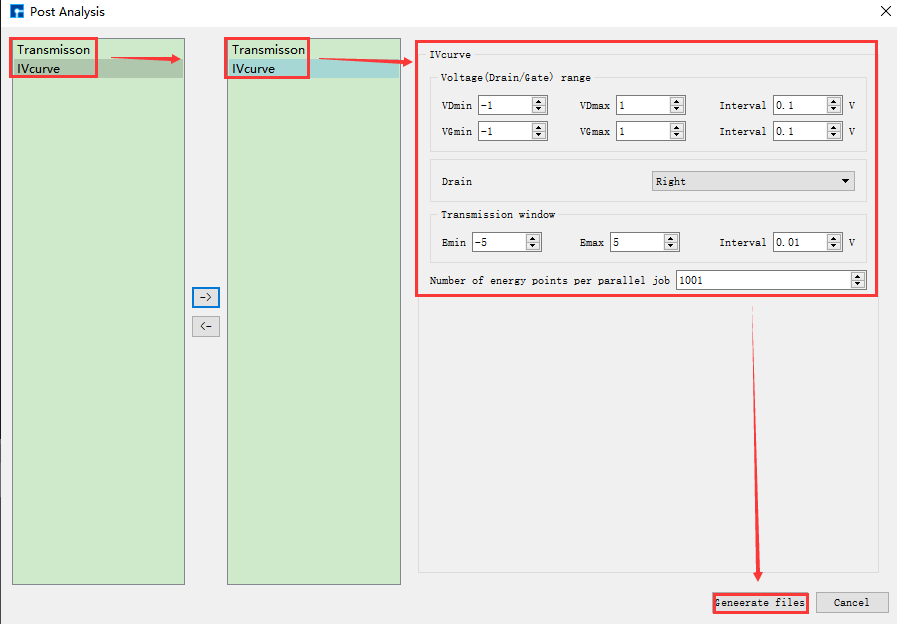
图 10.59 纯硅器件结构电流、电子透射谱输入文件参数设置界面¶
10.5.5. Nanoskim计算¶
10.5.5.1. Hamiltonian计算¶
生成Nanoskim计算各个输入文件后的Device Studio界面如 图 10.60 所示。以纯硅器件结构的Hamiltonian计算为例,在如 图 10.60 所示界面中,Device Studio的Project Explorer区域选中 Hamiltonian.input → 右击 → Run,弹出Run界面如 图 10.48 (Run界面)所示,点击界面中的 Run 按钮则可进行纯硅器件结构的Hamiltonian计算,在计算过程中,用户可在Device Studio的Job Manager区域查看计算状态。当计算任务处于排队中、计算中和计算完成时,Status 分别为Queued、Running、Finished。计算完成后,在Device Studio的Project Explorer区域选中 Hamiltonian.input → 右击 → Open Containing Folder 则找到 Hamiltonian.input 文件在本地电脑中的存储位置,并且可看到纯硅器件结构的Hamiltonian计算的结果文件 Hresult.mat,该文件在 IVSource 文件夹内。
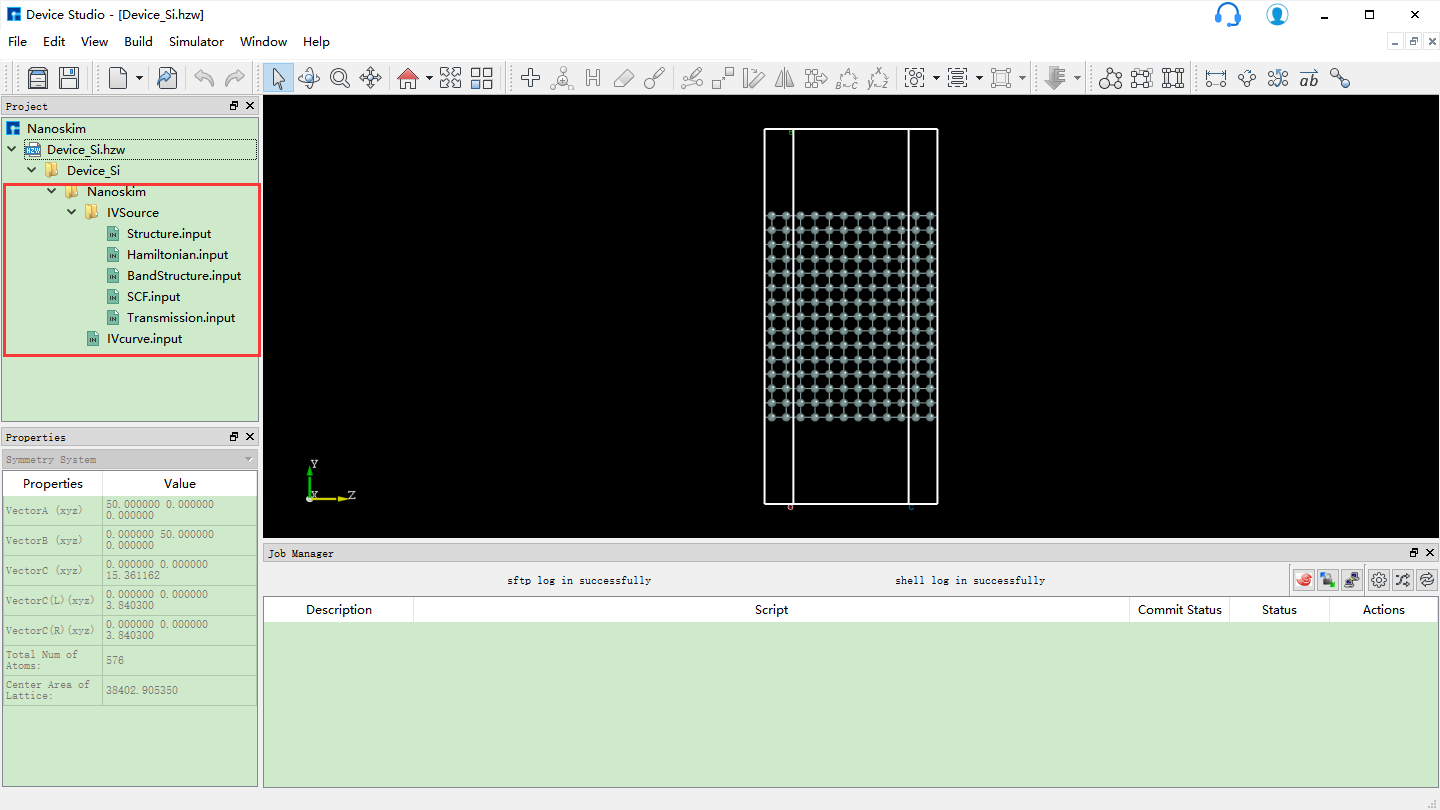
图 10.60 生成Nanoskim计算各个输入文件后的Device Studio界面¶
10.5.5.2. BandStructure计算¶
BandStructure计算需调用Hamiltonian计算的结果,故需先做Hamiltonian计算,再做BandStructure计算。本算例已经做过Hamiltonian计算,则直接调用Hamiltonian计算结果做BandStructure计算即可。在Device Studio的Project Explorer区域选中 Bandstructure.input → 右击 → Run,弹出Run界面如 图 10.48 Run界面 所示,点击界面中的 Run 按钮则可进行纯硅器件结构的能带计算。计算完成后,在Device Studio的Project Explorer区域选中 Bandstructure.input → 右击 → Open Containing Folder 则找到 Bandstructure.input 文件在本地电脑中的存储位置,并且可看到纯硅器件结构的能带计算的结果文件 BSresult.mat 和 BSresult.txt ,这2个文件都在 IVSource 文件夹内。
备注
Nanoskim的计算较为复杂,顺序为 Hamiltonian.input → Bandstructure.input → IVcurve.input,IVcurve.input 文件运行完成后,会在与IVSource文件夹同一目录下生成一系列 VD=##_VG=## 文件夹。如 VD=0.1__VG=0 文件夹下的一系列输入文件如下图所示,对于该文件夹下的各输入文件的运行这里不做详细说明,用户可参考 Nanoskim 快速使用说明手册,或发送邮件到邮箱 support@hzwtech.com 咨询。
10.5.6. Nanoskim计算结果的可视化分析¶
在如 图 10.60 所示界面(即:Device Studio图形界面)的Project Explorer区域选中 BSresult.txt → 右击 → Show View,则弹出 Nanoskim 能带的可视化分析界面如 图 10.61 所示。用户可根据需要调节坐标轴的取值范围;可通过滚动鼠标中键将可视化分析结果放大或缩小;可根据需要编辑坐标轴标注和标题,选择字体的大小和类型。选中如 图 10.60 所示界面中 Export 快捷图标,弹出导出可视化分析结果的图形界面,用户可根据需要设置导出图片的大小,选择图片的保存路径和保存格式,并给所保存的图片命名。
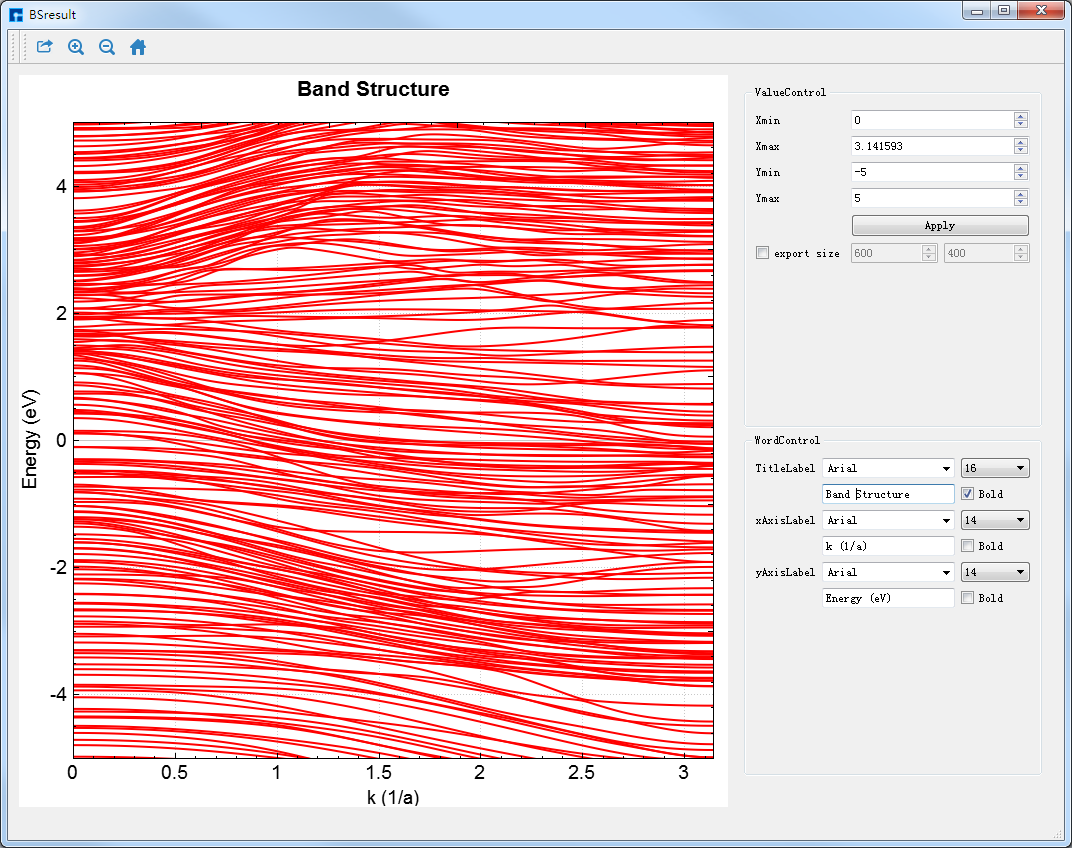
图 10.61 Nanoskim能带的可视化分析界面¶
亦可在Device Studio图形界面中选中 Simulator → Nanoskim → Analysis Plot,弹出导入能带结果文件的图形界面,在界面中找到 BSresult.txt 文件并选中,点击 打开 按钮即可弹出 Nanoskim 能带的可视化分析界面。
10.6. STEMS 实例¶
STEMS (Simulation Tools for the Evolution of Micro-Strucure)是一款基于相场或晶体相场理论方法 (Phase-Field /Phase-Field-Crystal Methods)的材料微观组织结构演化模拟软件。其中相场方法部分适用于研究多种合金凝固过程 中的组织结构演化过程,如支晶生长,共晶生长等现象。晶体相场部分用于研究晚期凝固过程中和凝固压力相关的纳米尺度机制,可以弥补相场方法在描述凝固缺陷方面的不足。
相场或晶体相场方法具有坚实的物理基础。相场方法通过求解一组描述凝固过程中固液界面移动过程的经典偏微分方程来研究材料在凝固过程中的微观组织结构演化。晶体相场方法通过求解基于变分方法和原子密度泛函的偏微分方程组来研究、模拟、理解和预测凝固过程中在纳米尺度发生的材料微组织演化。
STEMS 基于最新的相场和晶体相场方法,由领域内的世界著名科学家团队领衔开发,是目前唯一结合了相场和晶体相场方法,能够研究凝固过程中的晶体生长和缺陷生成机制等重要问题的模拟软件。相场部分使用新的巨势多相场模型,便于耦合多种合金的热力学自由能数据,可以研究快速凝固过程。
STEMS 可模拟的过程如下:凝固过程中的枝晶生长现象、共晶生长现象、定向凝固、快速凝固、凝固缺陷、凝固压力。

10.6.2. STEMS创建项目¶
双击Device Studio图标快捷方式,登录并启动Device Studio,在创建或打开项目界面中(启动软件后选择创建或打开项目的图形界面),根据界面提示选择创建一个新的项目( Create a new Project )或打开一个已经存在的项目( Open an existing Project )的按钮,选中之后点击界面中的 OK 按钮即可。若选择创建一个新的项目,用户可根据需要给该项目命名,如本项目命名为 STEMS,或采用软件默认项目名。
10.6.3. STEMS导入文件¶
在Device Studio的图形界面点击 File → Import → Import Local
,则弹出导入STEMS文件的界面如 图 10.63 所示,根据界面提示找到 PF-PFC.hzw 文件的位置,选中 PF-PFC.hzw 文件,点击 打开 按钮则导入 PF-PFC.hzw 文件后的Device Studio界面如 图 10.64 所示。在Device Studio中导入结构的其他方法这里不做详细说明,用户可参照 导入结构 节内容。
图 10.63 STEMS导入文件的界面¶
图 10.64 导入 PF-PFC.hzw 文件后的Device Studio界面¶
10.6.4. PFC计算输入文件的生成¶
在如 图 10.64 所示界面中选中 Simulator → STEMS → PFC,
弹出PFC参数设置界面如 图 10.65 所示,用户可根据计算需要分别选中界面中的 Init 、
Func、Run 分别设置参数如 图 10.65 、 图 10.66 和 图 10.67 所示,
之后点击界面中的 Generate files 即可生成PFC计算输入文件 Init.param 、Func.param 和 Run.Param,生成PFC计算输入文件后
的Device Studio图形界面如 图 10.68 所示。
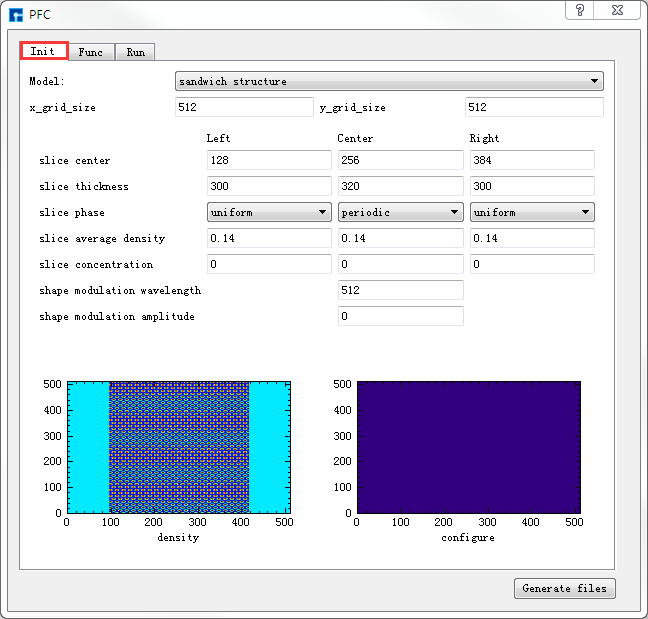
图 10.65 Init参数设置界面¶
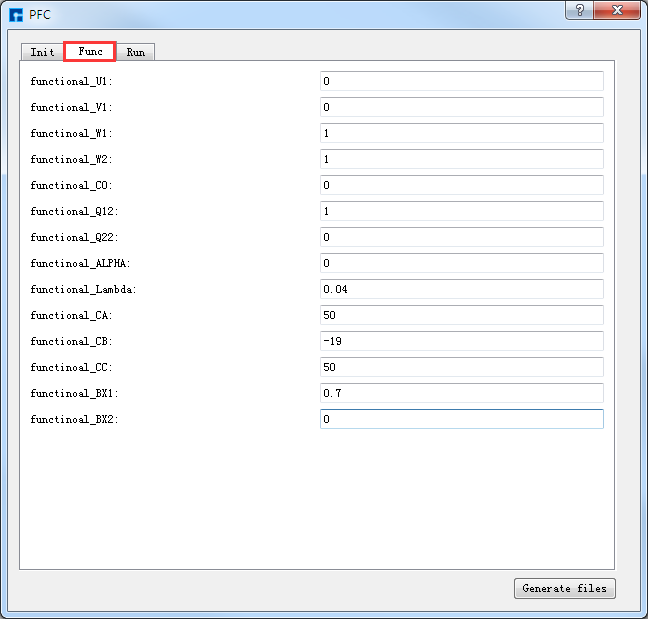
图 10.66 Func参数设置界面¶
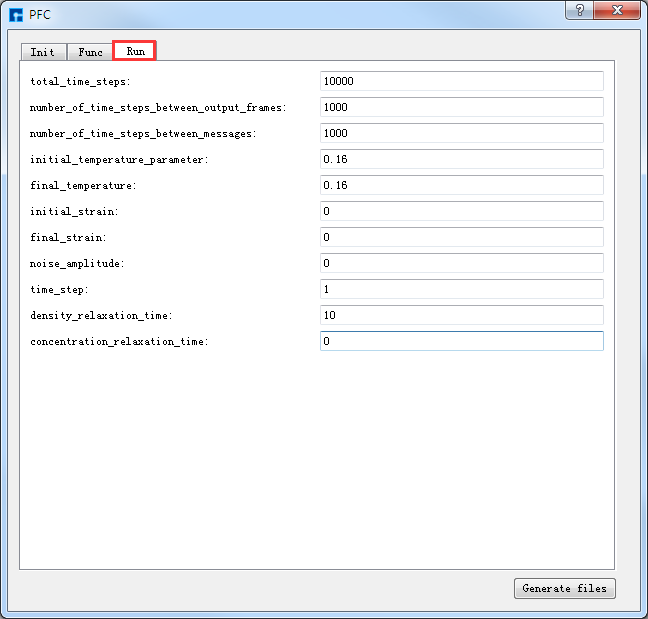
图 10.67 Run参数设置界面¶
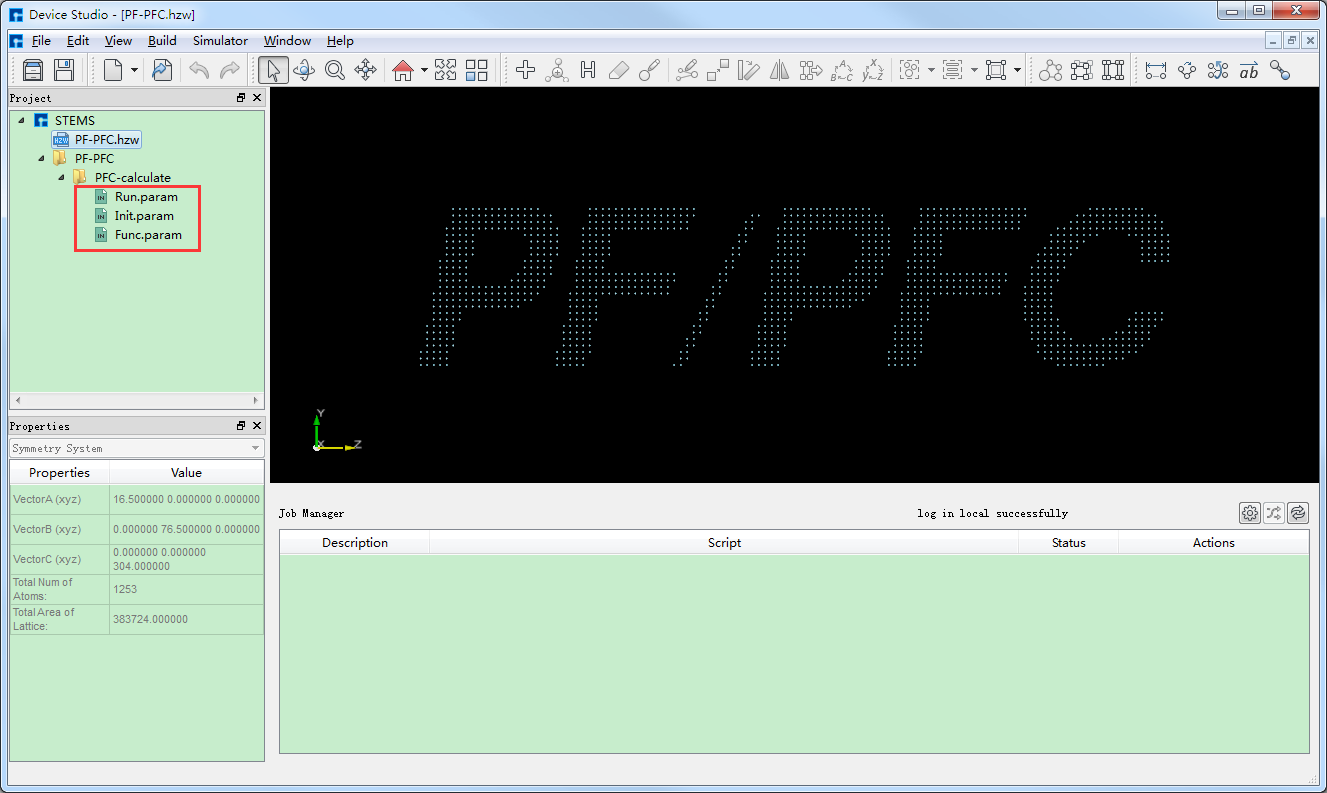
图 10.68 生成PFC计算输入文件后的Device Studio界面¶
10.6.5. PFC计算¶
在做PFC计算之前,需连接装有STEMS的服务器,具体连接过程这里不做详细说明,用户可参考 Nanodcal连接服务器 节内容。
连接好装有STEMS的服务器,在做PFC计算之前,用户可根据需要打开输入文件并查看文件中的参数设置是否合理,若不合理,则可选
择直接在文件中进行编辑或重新生成,最后再进行PFC计算。如打开 Init.param 文件,在 图 10.68 所示的界面中,
选中 Init.param → 右击 → Open with 即可查看到 Init.param 文件如下所示。对于其他输入文件,
用户可根据需要选择是否打开查看,这里不做详细说明。
initial_model = 3
x_grid_size = 512
y_grid_size = 512
slice_center_left = 128
slice_thickness_left = 300
slice_phase_left = 1
slice_average_density_left = 0.14
slice_concentration_left = 0
slice_center_center = 256
slice_thickness_center = 320
slice_phase_center = 0
slice_average_density_center = 0.14
slice_concentration_center = 0
shape_modulation_wavelength = 512
shape_modulation_amplitude = 0
slice_center_right = 384
slice_thickness_right = 300
slice_phase_right = 1
slice_average_density_right = 0.14
slice_concentration_right = 0
备注
在实际计算过程中,用户可通过 STEMS 使用教程 了解各参数的详细意义,从而根据计算需要合理设置参数。若想详细了解 STEMS ,可发送邮件到邮箱 support@hzwtech.com 咨询。
在如 图 10.68 所示的界面的Project Explorer区域中,选中 Run.param → 右击 → Run,弹出Run界面如 图 10.69 所示,根据计算需要设置参数后点击 Save 按钮保存相应的脚本,之后点击如 图 10.69 所示界面中
的 Run 按钮则可做STEMS的PFC计算。用户可在Job Manager区域中观察PFC计算状态,当PFC计算任务处于排队中、
计算中和计算完成时,Status 分别为Queued、Running、Finished;在计算过程中,点击 刷新 按钮(即 refresh 按钮),通过 Commit Status 、Status 可查看到PFC计算的计算进度和计算状态;计算完成后Device Studio的Job Manager区域如 图 10.70 所示,点击 Action 下的 下载 按钮弹出下载PFC计算结果界面如 图 10.71 所示,在该界面找到PFC计算的结果文件( PFC计算结果文件即后缀名为 .dat 的文件,如 d_mpi.0.dat ),点击 Download 则可下载,下载后可在软件的Project Explorer区域查看到该结果文件。
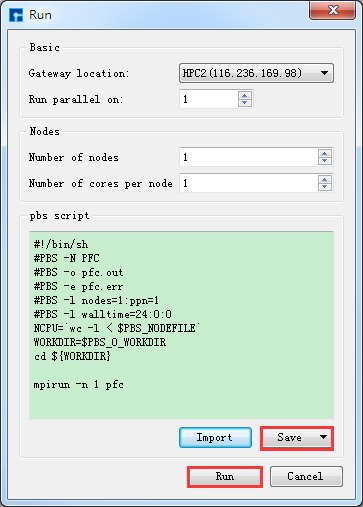
图 10.69 STEMS的Run界面¶
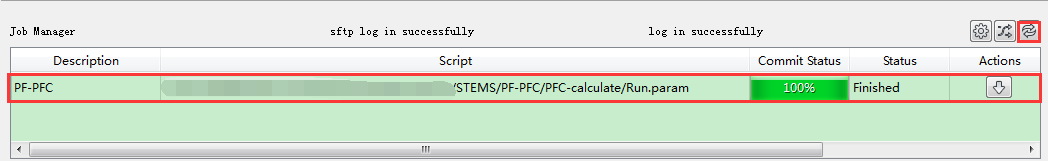
图 10.70 PFC计算完成后的Device Studio的Job Manager区域¶
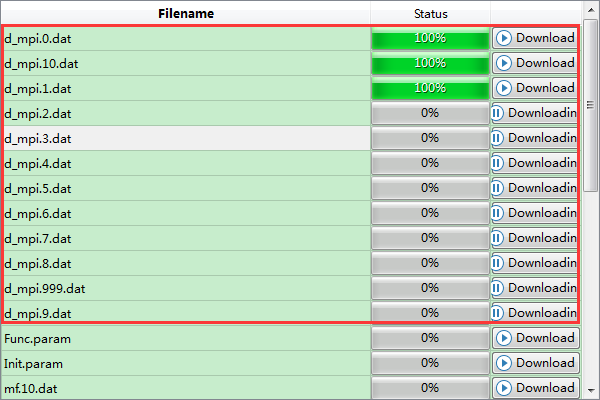
图 10.71 下载PFC计算结果的界面¶
PFC计算结果下载完成后的Device Studio界面如 图 10.72 所示。
图 10.72 PFC计算结果下载完成后的Device Studio界面¶
10.6.6. PFC计算结果的可视化分析¶
在如 图 10.72 所示界面中的Project Explorer区域内,选中任意一个PFC计算的结果文件,如 d_mpi.0.dat → 右击 → Open Containing Folder 则找到PFC计算结果文件在本地电脑中的存储位置如 图 10.73 所示。在如 图 10.72 所示的Device Studio图形界面中,点击
View → generate GIF,弹出generategif界面如 图 10.74 所示,在该界面中点击 File → import,弹出导入PFC计算结果文件的界面,按照提示将结果文件导入后,在generategif界面中点击 File → export,弹出将PFC计算结果的可视化分析结果导出的界面,用户可根据需要选择导出的位置,并给可视化分析结果命名,或采用默认命名,如命名为 PFC.gif ,PFC计算结果的可视化分析(即 PFC.gif 文件)如 图 10.75 所示。
图 10.73 PFC计算结果文件在本地电脑中的存储位置¶
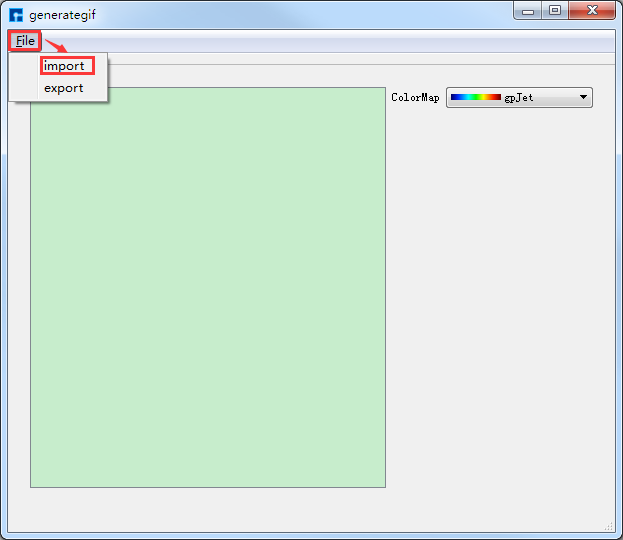
图 10.74 generategif界面¶

图 10.75 PFC计算结果的可视化分析(即 PFC.gif 文件)¶
10.7. MOMAP 实例¶
MOMAP ( Molecular Materials Property Prediction Package )是一款研究和设计有机分子材料发光和传输性能以及定量预测发光效率 和迁移率的软件。目前广泛应用于有机显示材料、有机照明材料、有机半导体材料、有机太阳能电池材料、有机光检测、生物传感材料、有机光通讯材料、有机光催化等领域。

与无机发光材料相比,有机发光材料具有很多优点:光程范围大、易得到蓝光、亮度大、效率高、驱动电压低、耗能少、具有特有的柔性、制作工艺简 单、来源丰富、易加工、可大面积制作等优势,在有机光电器件方面得到了广泛应用和快速发展。由于典型的有机发光体系的激发态衰减时间尺度从纳 秒级别直到微秒和毫秒,因此基于微扰理论的费米黄金规则成为处理复杂体系激发态衰减过程的最有效的理论方法之一。
以 azulene的吸收光谱、荧光光谱以及辐射速率的计算 为例详细描述 MOMAP 软件在Device Studio中的应用。
10.7.1. MOMAP软件功能结构¶
MOMAP的软件功能结构如 图 10.76 所示,能够根据其他量化软件计算得到分子基态、激发态信息,利用电子——振动耦合程序计算频率、模式位移、 Duschinsky转动矩阵,为接下来的计算做准备。MOMAP所需的分子基态、激发态信息主要包括:分子在基态以及激发态的平衡构型、能量、振动模式 以及两个电子态之间的跃迁偶极矩、非绝热耦合向量、自旋轨道耦合常数、转移积分等,这些信息可通过目前成熟的量化软件,如Gaussian、Turbomole、NWChem、Dalton、BDF等计算得到。
基于电子——振动耦合程序的结构,用户能够利用MOMAP内各种程序计算分子吸收光谱、荧光光谱、磷光光谱、辐射速率、内转换速率、系间窜越速率以 及电荷转移速率,并以此最终定量的得到分子的荧光、磷光的量子产率以及迁移率。
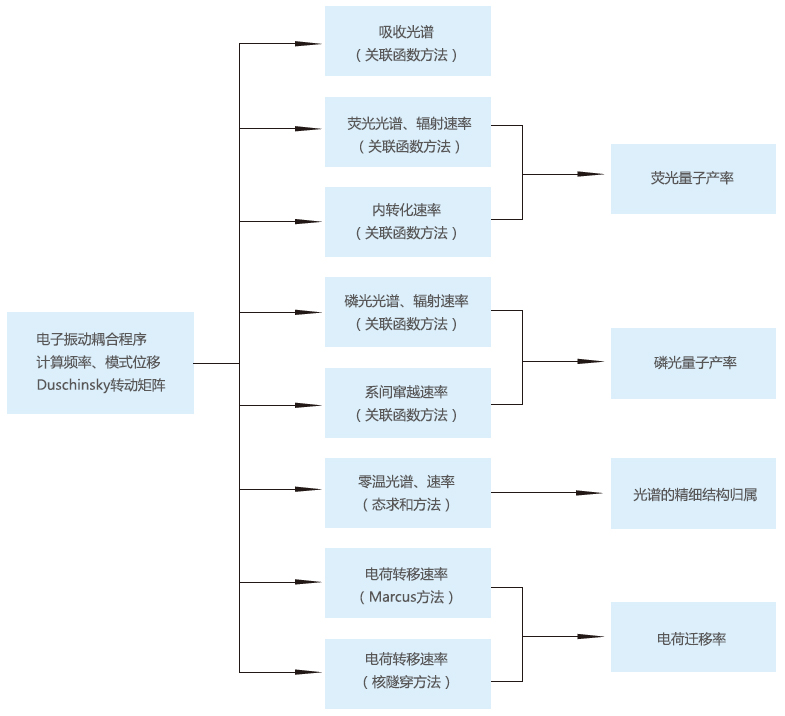
图 10.76 MOMAP软件功能结构图¶
10.7.2. BDF计算部分¶
BDF完成基态和激发态的优化任务后(opt+freq和TDDFT+opt+freq),收集基态和激发态的输出文件 azulene-s0.out 、 azulene-s1.out 和 azulene-s1.out.tmp。以及hess文件 azulene-s0.hess 和 azulene-s1.hess。再进行非绝热耦合计算任务,收集输出文件 azulene-nacme.out。
10.7.3. MOMAP计算部分¶
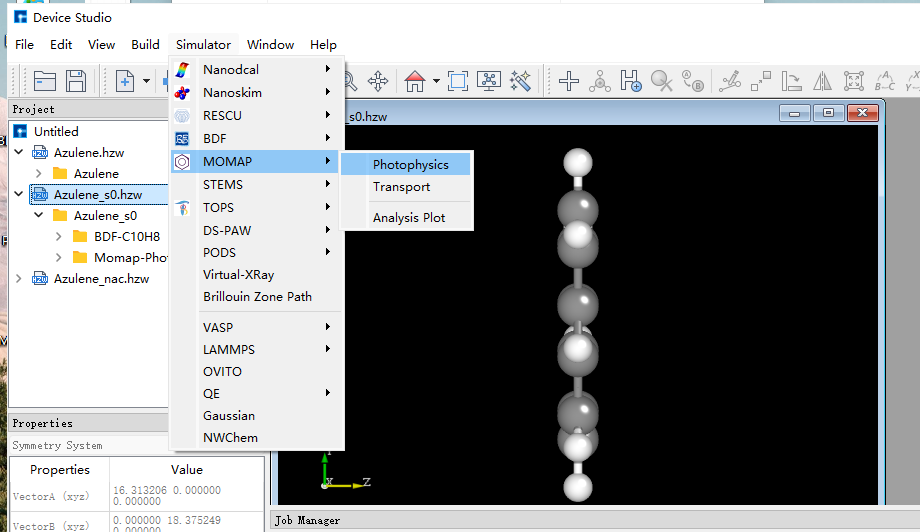
10.7.3.1. EVC计算¶
电子振动耦合(electron-vibration coupling, EVC)计算,该计算基于量化计算输出的分子振动频率、力常数矩阵,同时在内坐标以及直角坐标系下,计算分子跃迁发生初末态间的模式位移、黄昆因子、重整能以及Duschinsky转动矩阵。 双击基态结构,依次点击Simulator、MOMAP和Photophycisc进入MOMAP参数设置:
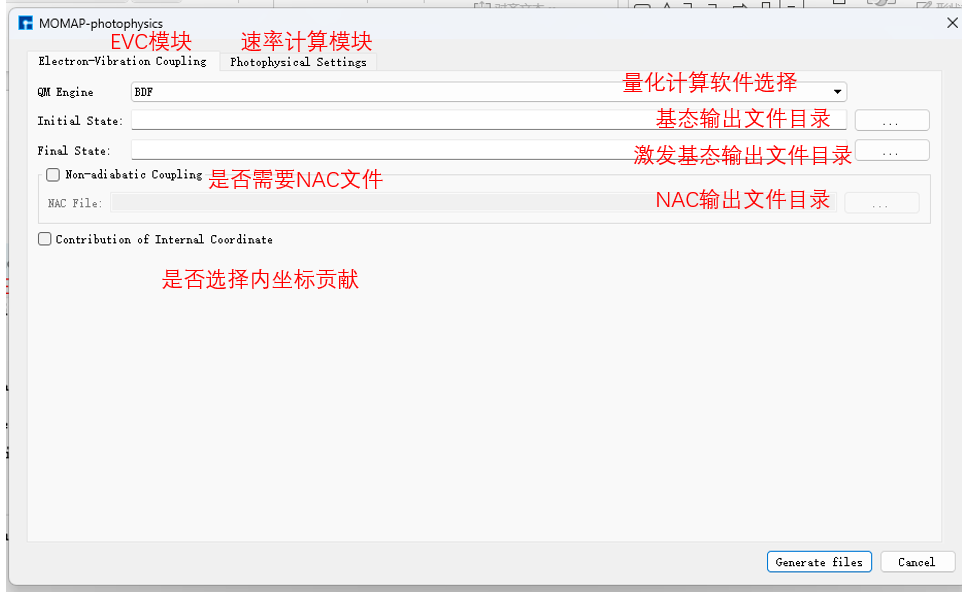
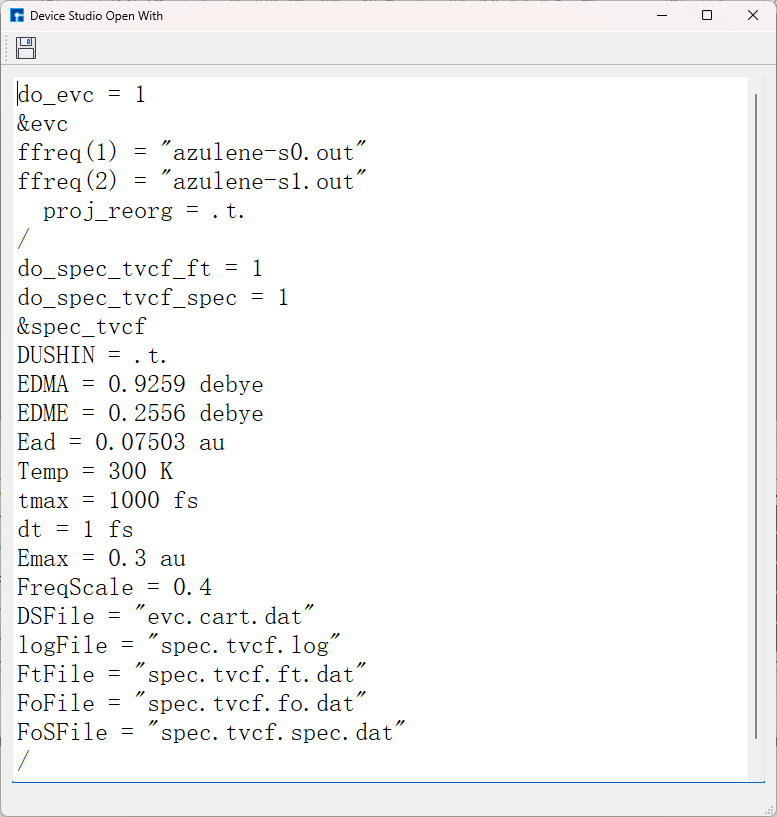
选定 azulene-s0.out 和 azulene-s1.out 路径后,点击Generate files生成MOMAP输入文件 momap.inp,选择 momap.inp 并点击右键,点击open with可查看 momap.inp:
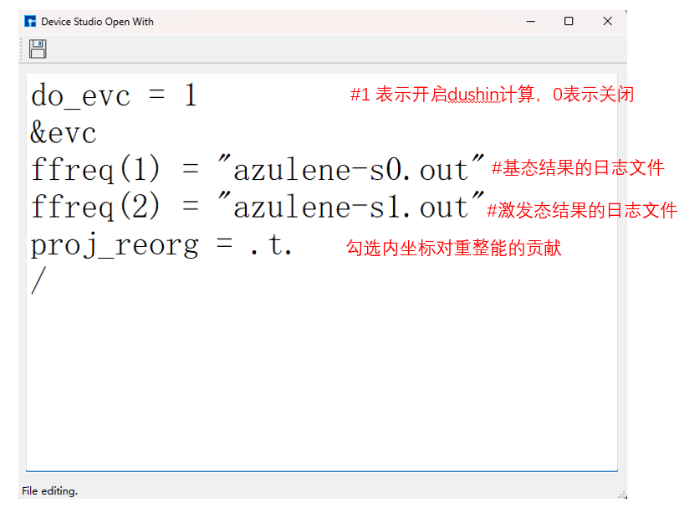
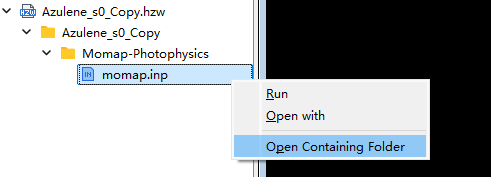
生成输入文件后 azulene-s0.out 和 azulene-s1.out 会直接复制到EVC计算文件夹中。按照下图选择 momap.inp 并右键点击,左击open Containing Folder打开EVC计算文件夹,会发现已经存在基态和激发态的输出文件,momap.slurm 为momap的提交脚本。将 azulene-s0.hess 和 azulene-s1.hess 文件复制至此文件夹后,即可点击run提交计算任务。
计算完成后任务栏Status下显示Completed,点击右侧的下载按钮——向下箭头:

双击文件即可下载。其中 evc.cart.dat 为利用直角坐标系计算得到的模式位移、黄昆因子、重整能以及 Duschinsky 转动矩阵的结果;而 evc.dint.dat 为内坐标计算模式位移、黄昆因子、重整能,直角坐标计算Duschinsky转动矩阵的结果。
Open with打开 evc.cart.dat 文件,查看总重组能的数值:
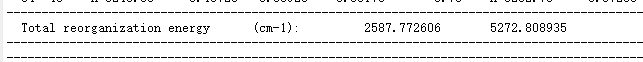
并与 evc.dint.dat 文件中的数值进行比较:
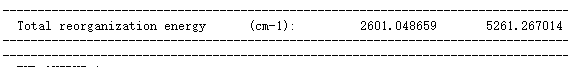
比较 evc.cart.dat 以及 evc.dint.dat 文件的重整能,若重组能相差不大( \(<1000 \, \text{cm}^{-1}\)),使用 evc.cart.dat 文件进行后续计算,若重整能相差较大(一般情况 evc.cart.dat 大于 evc.dint.dat ),使用 evc.dint.dat 文件进行后续计算。这里可使用 evc.cart.dat 进行下一步S1→S0的荧光辐射速率计算。
此外,还可以根据EVC计算的结果文件做更多的后处理。
evc.dx.x.com 和 evc.dx.x.xyz 为两个电子态分子叠加图,其中 evc.dx.x.com 可用GaussView打开,在View-Display Format-Molecule中选择Tube类型,显示如下:
evc.cart.abs 为Duschinsky矩阵文件,可用来画Duschinsky矩阵二维图。可以在Device Studio中,选择Simulator-momap-analysis,打开 evc.cart.abs 文件,显示如下:
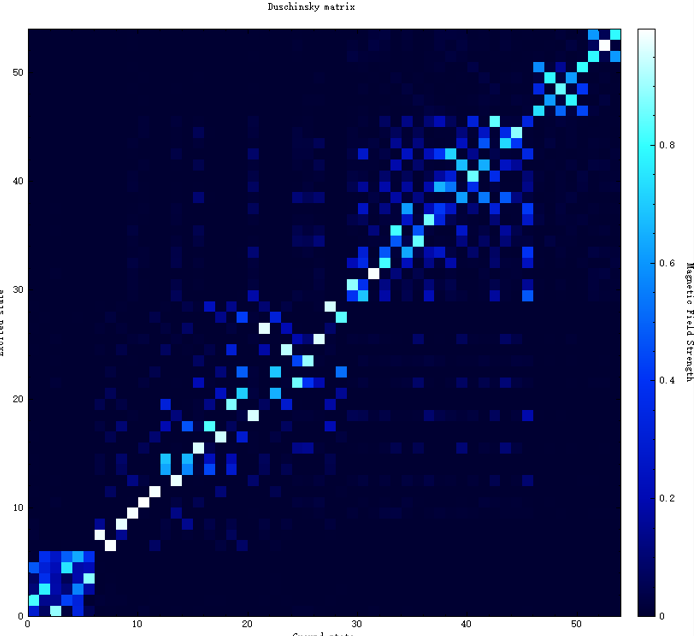
10.7.3.2. 荧光光谱及荧光辐射速率的计算¶
接下来进行S1→S0的荧光光谱及荧光辐射速率的计算。首先需要计算绝热激发能和吸收跃迁偶极矩(Electronic Transition Dipole Absorption, EDMA)以及发射跃迁偶极矩(Electronic ransition Dipole Emission,EDME)。
选择 azulene-s0.out,右击open with containing folder,打开 azulene-s0.out 。从文件开始向下查找字段“Electronic total energy”, 其后即为基态能量,\(E_{s0} = -385.878076 \, \text{Hartree}\) 。

选择 azulene-s1.out.tmp,右击open with containing folder,打开 azulene-s0.out.tmp。搜索字段“Statistics for [dvdson_rpa_block]: ”,读取No.1态的能量 ,\(E_{s1} = -385.803048 \, \text{a.u.}\),即为S1态的单点能。
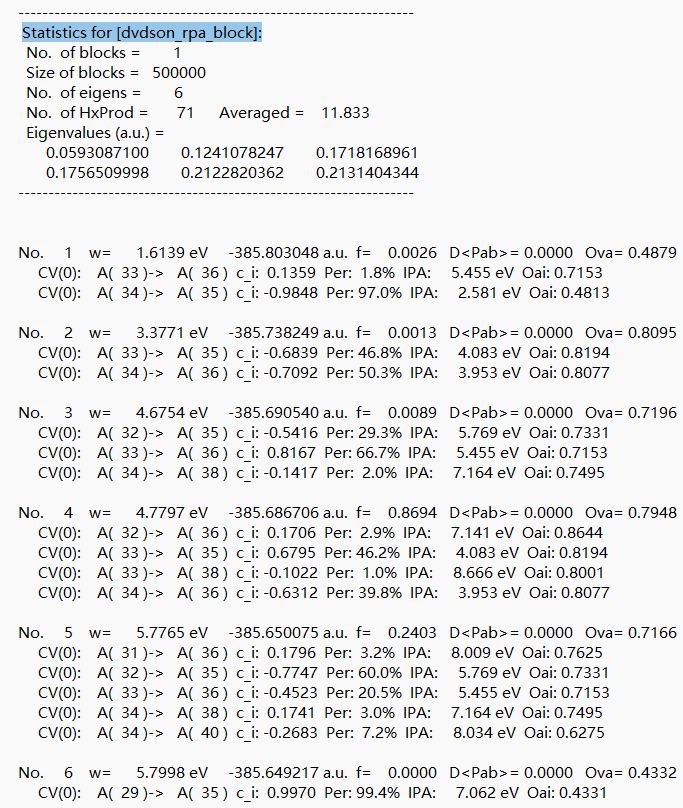
得到绝热激发能 \(E_{ad}\)
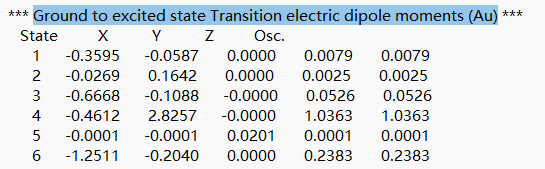
从 azulene-s1.out.tmp 文件开始向下查找第一个tddft模块的内容,字段“Ground to excited state Transition electric dipole moments (Au)” 下的State 1的跃迁电偶极矩,可以得到momap需要的参数EDMA。
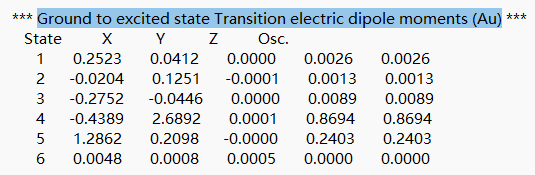
在文件末尾向上查找第一个tddft计算模块module tddft 。根据tddft模块的 “Ground to excited state Transition electric dipole moments (Au) ”中的State 1的跃迁电偶极矩,得到momap需要的参数EDME。
上述绝 \(E_{ad}\)、EDMA和EDME计算完成即可开始荧光光谱及荧光辐射速率的计算。此部分计算需要EVC计算所得的evc.*.dat 文件作为输入。和EVC一样,双击基态结构,依次点击Simulator、MOMAP和Photophycisc进入MOMAP参数设置,选定 azulene-s0.out 和 azulene-s1.out 路径后,点击Photophysical Settings进入光谱计算参数设定界面,选择荧光辐射速率项目,填入绝热激发能和吸收、发生跃迁偶极矩,并设置好其他参数:
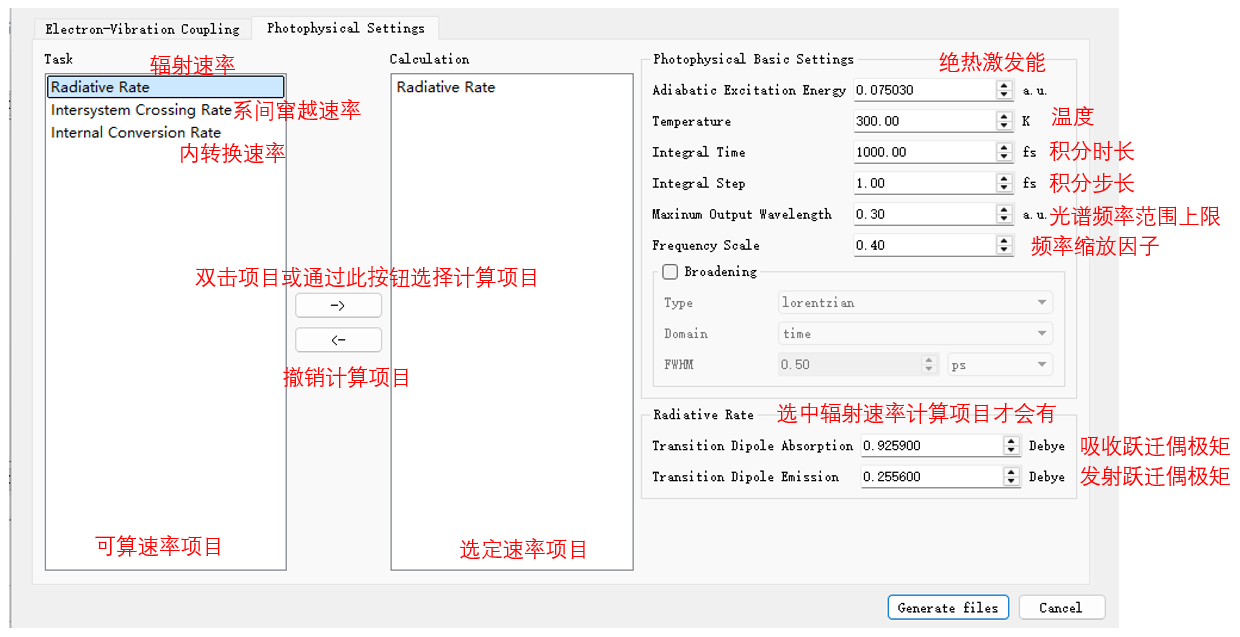
选择 momap.inp 并点击右键,点击open with可查看到 momap.inp
选择 momap.inp 并点击右键,点击run即可提交计算任务,进行荧光光谱及荧光辐射速率的计算。计算完成后主要有以下文件输出:
spec.tvcf.fo.dat #谱函数输出文件
spec.tvcf.ft.dat #关联函数输出文件
spec.tvcf.log #log 文件
spec.tvcf.spec.dat #光谱文件
计算完成后先确认关联函数是否收敛,将 spec.tvcf.ft.dat 拖入origin中,选择第一列和第二列作图:
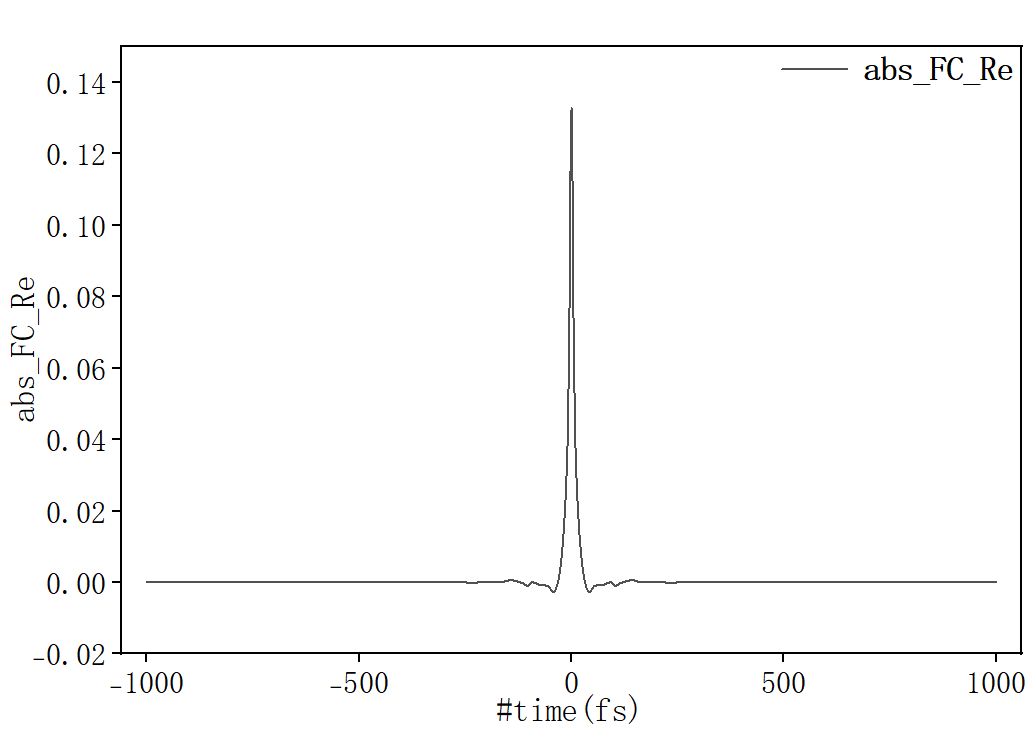
随时间趋于0表示吸收光谱计算收敛。选择第一列和第四列作图:
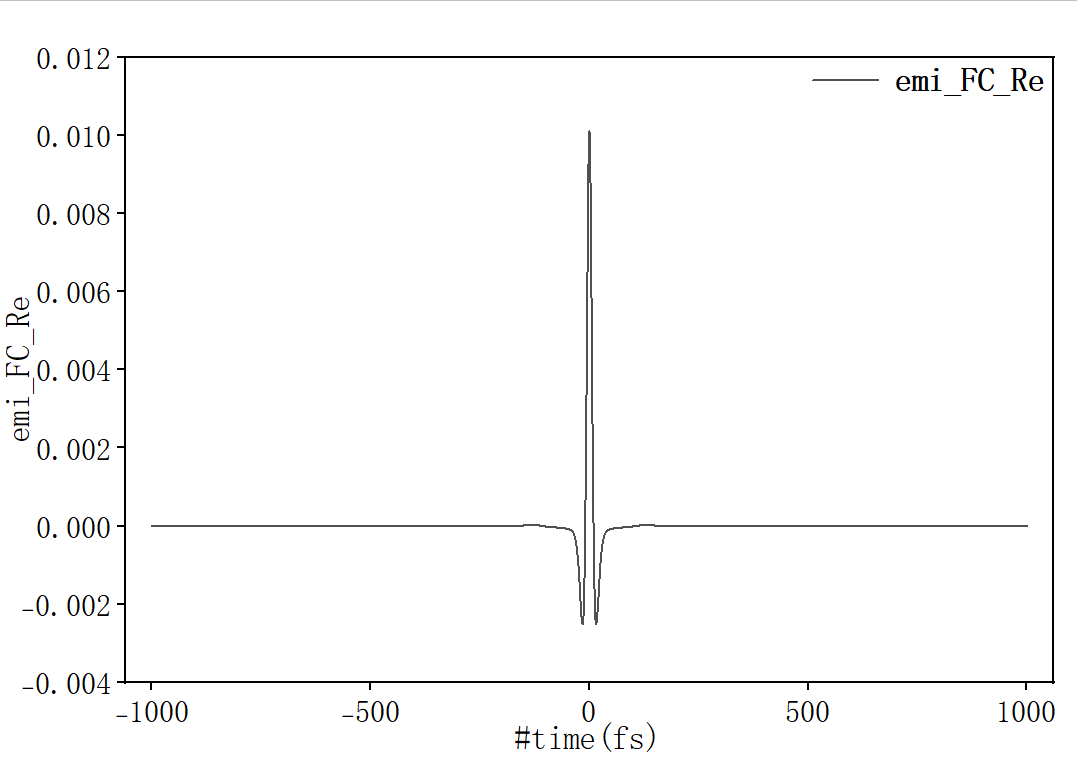
随时间趋于0表示发射光谱计算收敛。
确认关联函数收敛后,根据光谱文件 spec.tvcf.spec.dat,将 spec.tvcf.spec.dat 拖入origin中,选择第三列和第五列作图,得到吸收光谱:
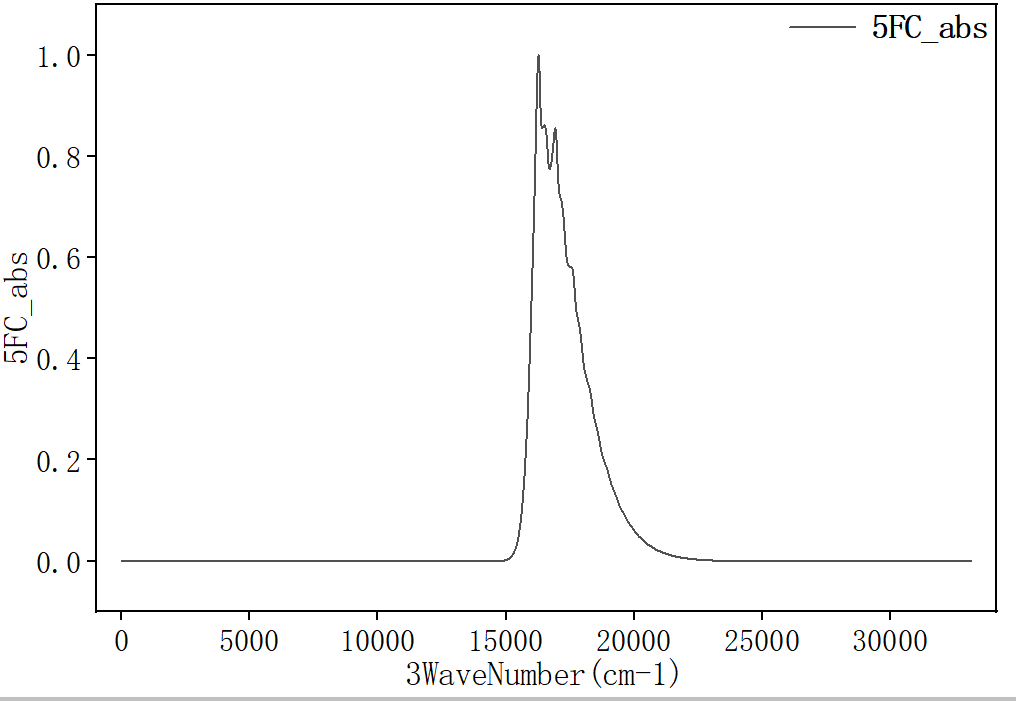
选择第三列和第六列作图,得到发射光谱:
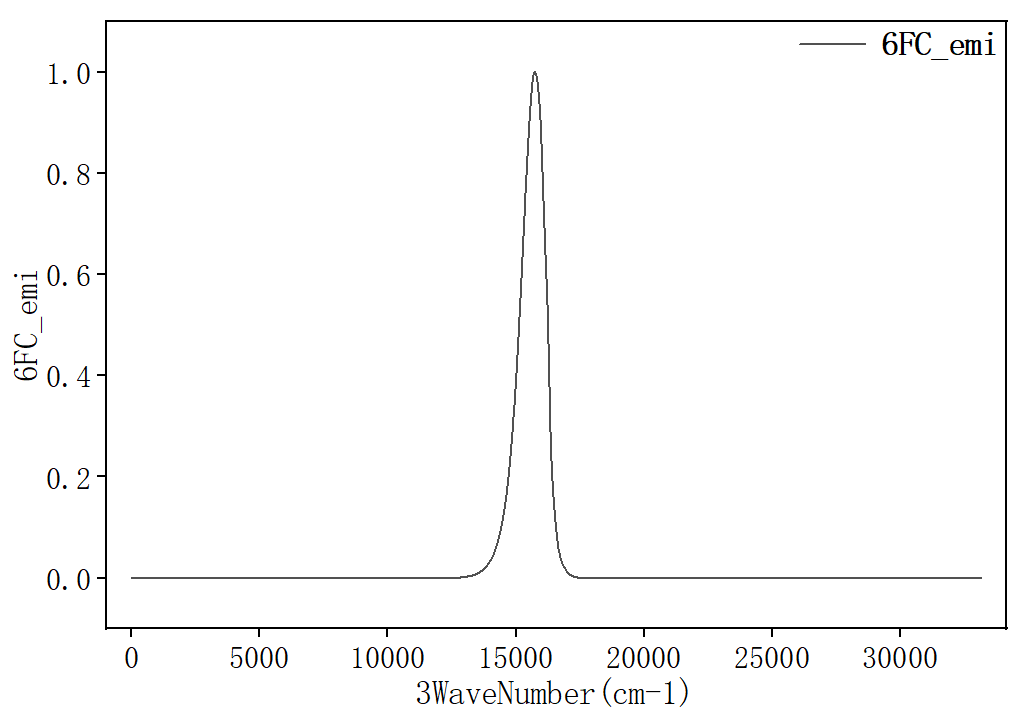
辐射速率kr可在 spec.tvcf.log 文件末端读取。如下图所示,radiative rate第一个数值和第二个数值都表示辐射速率,单位分别是 \(\text{a.u.}\) 和 \(\text{s}^{-1}\),第三个数值表示寿命。计算得到该分子的荧光辐射速率kr为 \(7.68731507×10^{4} \, \text{s}^{-1}\) 。

10.7.3.3. 内转换速率¶
内转换速率的EVC振动分析计算需要非绝热耦合计算结果文件。第一步为EVC振动分析计算。将非绝热耦合计算结果文件改名为 azulene-nacme.out,将 azulene-s0.out 和 azulene-s1.out,以及 azulene-s0.hess 和 azulene-s1.hess 放在内转换速率计算文件夹下,以下操作选择基态、激发态和NAC结果的路径后,生成momap输入文件。
选择 momap.inp 并点击右键,点击open with可查看到 momap.inp:
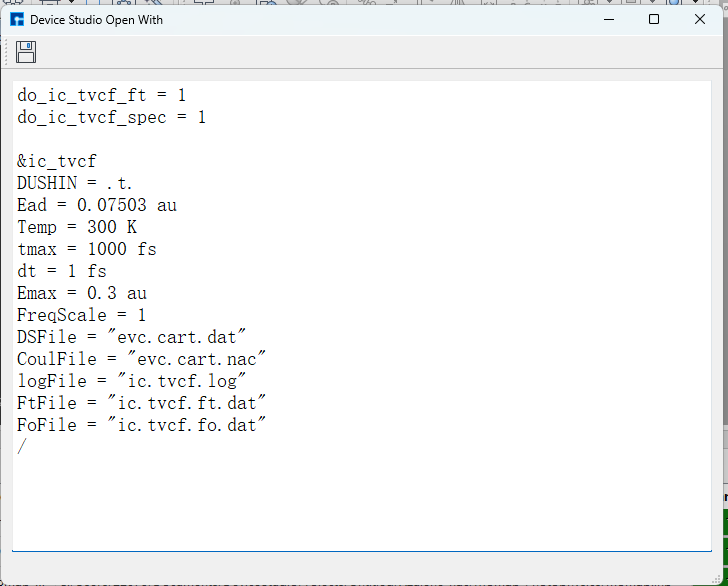
与前述荧光辐射速率的 evc 计算相比,多出一个 evc.cart.nac 文件,该文件将和 evc.cart.dat 文件一起用于接下来的内转换速率的计算。选择Internal Conversion Rate项目内转换速率计算,参数设置如下:
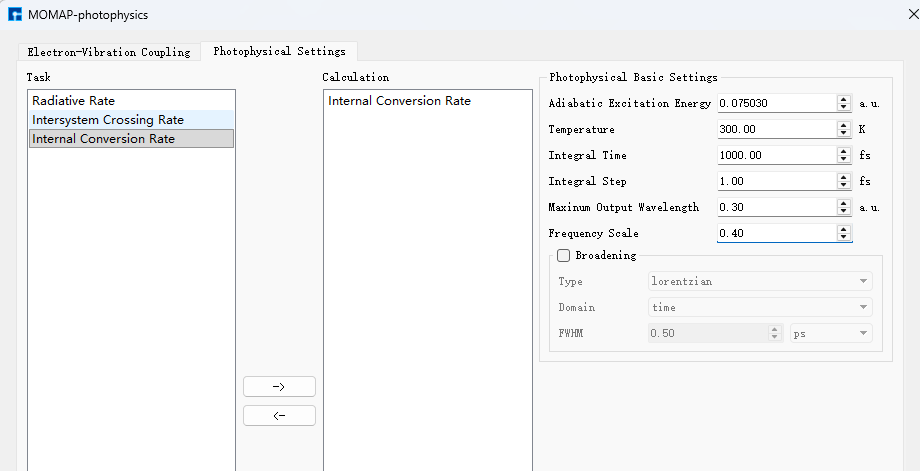
选择 momap.inp 并点击右键,点击open with可查看到 momap.inp:
其中 ic.tvcf.ft.dat 为关联函数文件, 计算结束后首先确认关联函数是否收敛,将 ic.tvcf.ft.dat 拖入origin中,选择第一列和第二列作图:
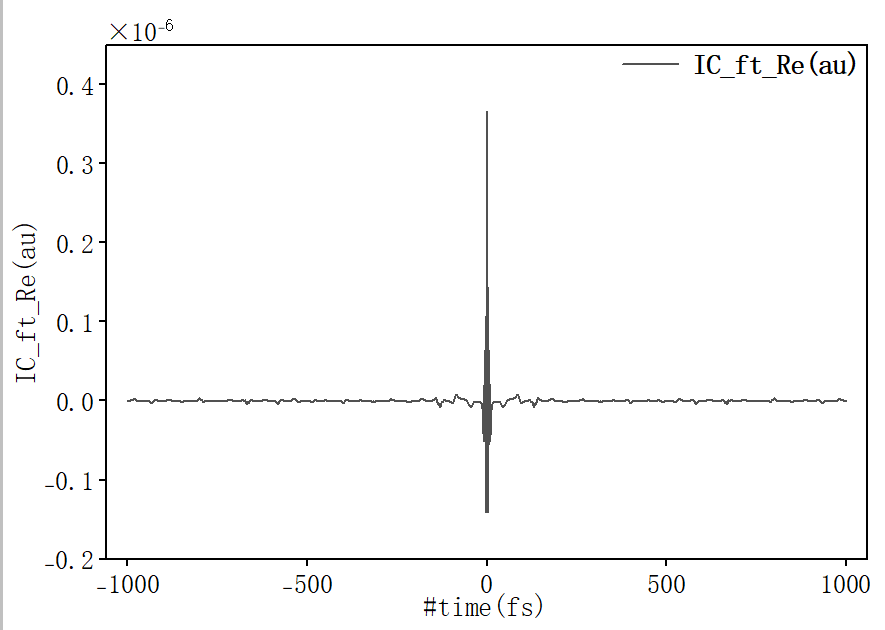
随时间趋于0表示关联函数收敛。其中 ic.tvcf.fo.dat 为谱函数文件,为检查是否满足能隙定律,将 ic.tvcf.fo.dat 拖入origin中,选择第三列和第七列作图
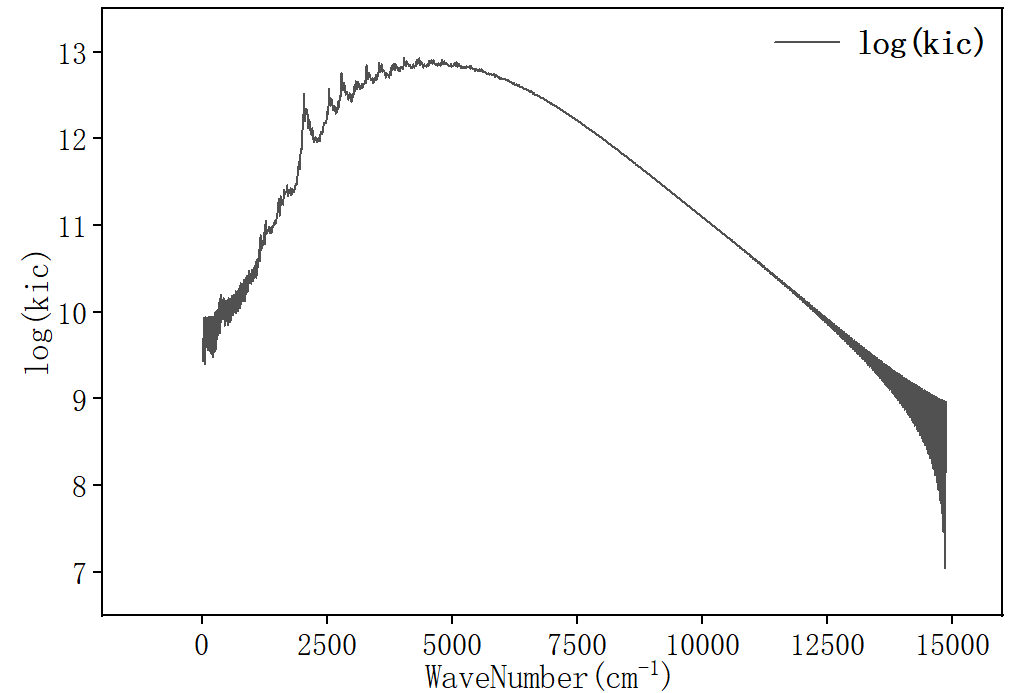
此外,ic.tvcf.fo.dat 文件中第一列和第六列表示不同Ead下的非辐射速率。
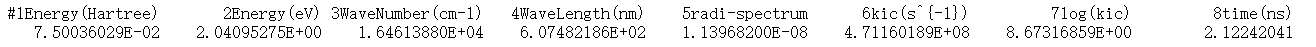
这里S1→S0的非辐射速率为2.70536301E+8。
10.8. VASP 实例¶
VASP (Vienna Ab-inito Simulation Package)是维也纳大学Hafner小组开发的进行电子结构计算和量子力学-分子动力学模拟软件包。它是目前材料模拟和计算物质科学研究中十分流行的商用软件之一。VASP 使用平面波基组,电子与离子间的相互作用使用模守恒赝势(NCPP)、超软赝势(USPP)或投影扩充波(PAW)方法描述。VASP 软件作为目前国内国际上权威的第一性原理计算软件,可以研究多种体系,包括金属及其氧化物、半导体、晶体、掺杂体系、纳米材料、分子、团簇、表面体系和界面体系等。
VASP不仅能够计算得到各种体系的平衡结构和能量,而且还能够对材料的电子性质进行精确的预测,深度剖析材料的各种理化性质。VASP 软件功能强大,性能稳定,具有非常高效的计算效率,可以使用较小的内存实现大规模的高效率并行计算,是目前做固体材料第一性原理计算效率很高的商用软件之一。
VASP软件官网:详见 https://cms.mpi.univie.ac.at/vasp/vasp/vasp.html
鸿之微科技(上海)股份有限公司在 Device Studio 2020B 中开发了适用于第一性原理科学计算软件 VASP 的计算模块。使用Device Studio,用户可在其图形界面中方便快捷的搭建或导入计算所需的结构,并可在结构3D显示区域查看其结构的3D视图。搭建好结构后用户可在 VASP 计算模块,根据计算需要,在简洁友好的界面中设置参数 生成计算所需的输入文件,之后连接装有VASP的远程服务器进行相关计算,在计算过程中可 实时监测任务的计算状态,计算完成后可 对VASP的计算结果进行可视化分析。
目前用户可通过Device Studio生成 VASP 以下计算输入文件的生成: 自洽、能带、态密度、AIMD、NEB、结构弛豫;支持 VASP 的 能带、投影能带、态密度、投影态密度、电荷密度CHGCAR、势函数LOCPOT 等计算结果的可视化分析。
以 GaSe晶体结构的结构弛豫计算 为例来详细描述 VASP 在Device Studio中的应用。
备注
Device Studio仅提供VASP软件使用接口,VASP软件自身相关版权请用户自行负责。

10.8.2. VASP创建项目¶
双击Device Studio图标快捷方式,登录并启动Device Studio,在创建或打开项目界面中(启动软件后选择创建或打开项目的图形界面),根据界面提示选择创建一个新的项目( Create a new Project )或打开一个已经存在的项目( Open an existing Project )的按钮,选中之后点击界面中的 OK 按钮即可。若选择创建一个新的项目,用户可根据需要给该项目命名,如本项目命名为 VASP,或采用软件默认项目名。
10.8.3. VASP导入结构¶
在Device Studio的图形界面中点击 File → Import → Import Local,则弹出导入VASP结构文件的界面如 图 10.78 所示,根据界面提示找到 GaSe.hzw 结构文件的位置,选中 GaSe.hzw 结构文件,点击 打开 按钮则导入 GaSe.hzw 结构后的Device Studio界面如 图 10.79 所示。在Device Studio中导入结构的其他方法这里不做详细说明,用户可参照 导入结构 节内容。
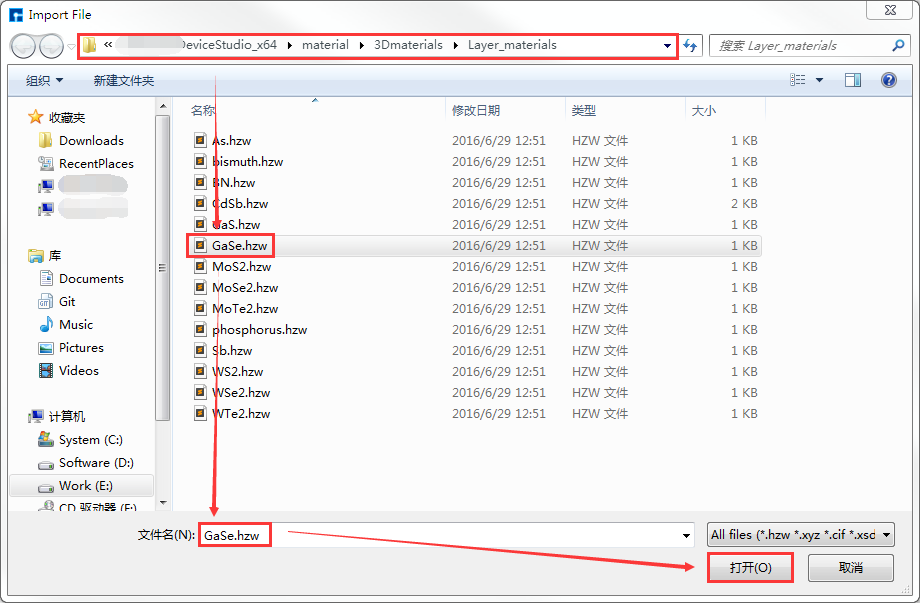
图 10.78 导入结构文件的界面¶
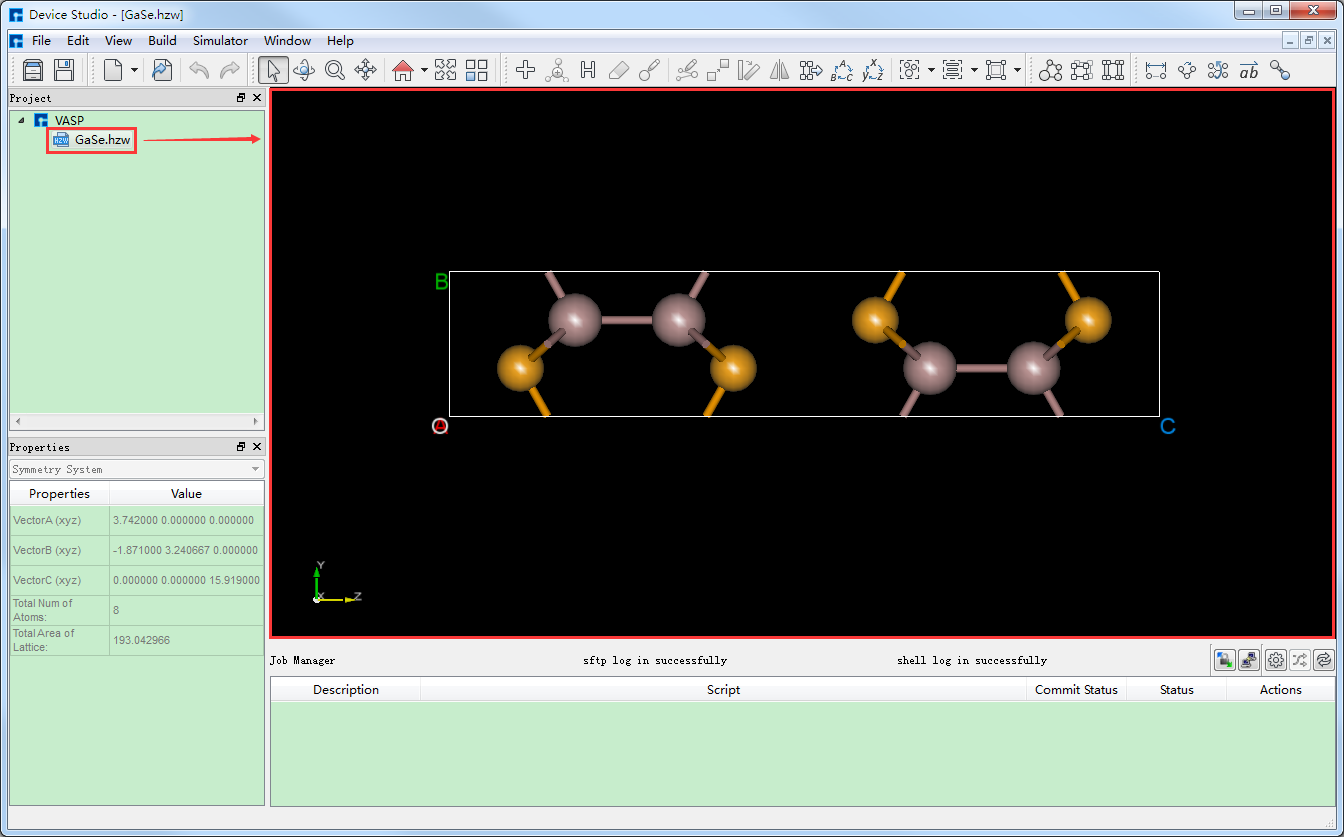
图 10.79 导入 GaSe.hzw 结构后的Device Studio界面¶
10.8.4. VASP输入文件的生成¶
VASP计算需要用到VASP的赝势文件,故在生成输入文件之前,可先在Device Studio中设置VASP赝势文件的调用路径。在Device Studio的图形界面中点击 File → Options,则弹出Options界面如 图 10.80 所示,在Options界面①处点击 Browse 按钮,则弹出Open Directory界面,用户可根据界面指示找到VASP的赝势文件并导入,导入VASP赝势文件后,则Options界面中的②处显示VASP赝势文件的调用路径。
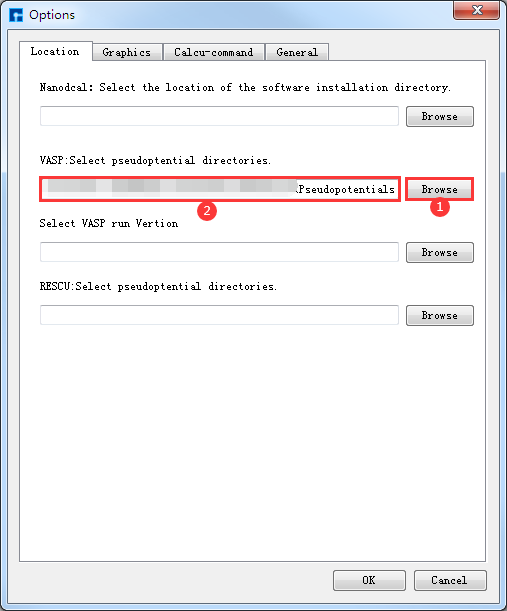
图 10.80 Device Studio的Options界面¶
以生成 GaSe晶体结构的结构弛豫计算 的输入文件为例,在如 图 10.79 所示Device Studio图形界面中,选中 Simulator → VASP →
Relaxation,弹出VASPRelaxation界面。根据计算需要分别选中VASPRelaxation界面中 Relaxation、Basic setting、
Advanced 设置参数分别如 图 10.81 、 图 10.82 和 图 10.83 所示,设置好参数后,可分别点击界面中的 INCAR、KPOINTS、
POSCAR、POTCAR 预览生成的GaSe晶体结构的结构弛豫计算的输入文件INCAR、KPOINTS、POSCAR、POTCAR分别如 图 10.84 、 图 10.85 、 图 10.86 和 图 10.87 所示,之后点击界面中的 Generate files 即可生成GaSe结构弛豫计算的输入文件 INCAR、KPOINTS、POSCAR、POTCAR。
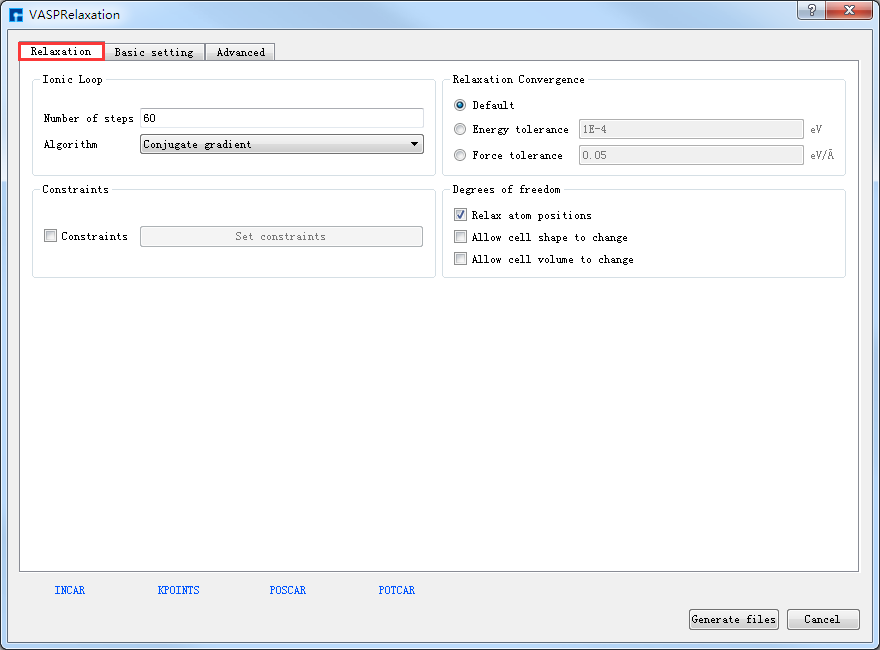
图 10.81 Relaxation参数设置界面¶
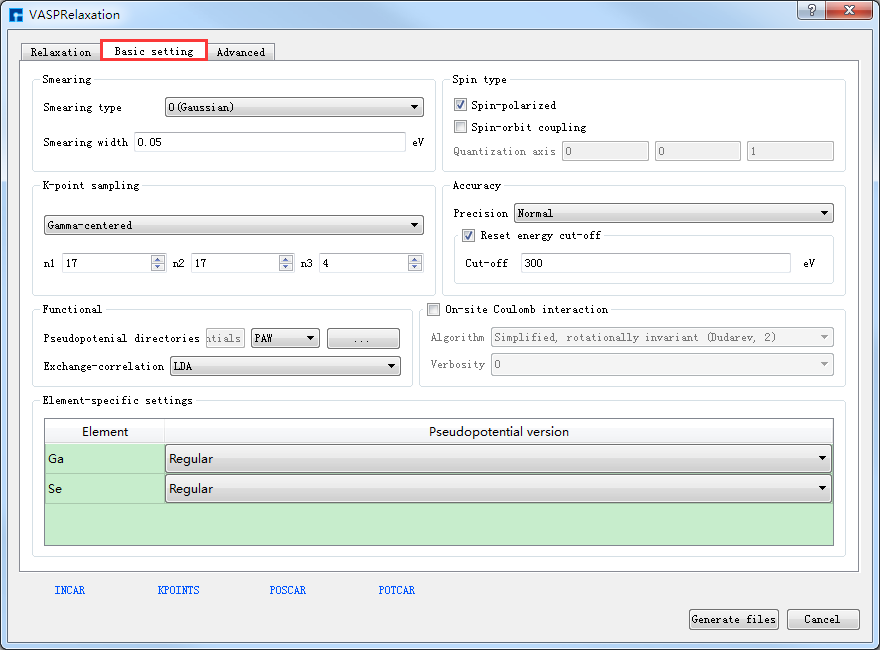
图 10.82 Basic setting参数设置界面¶
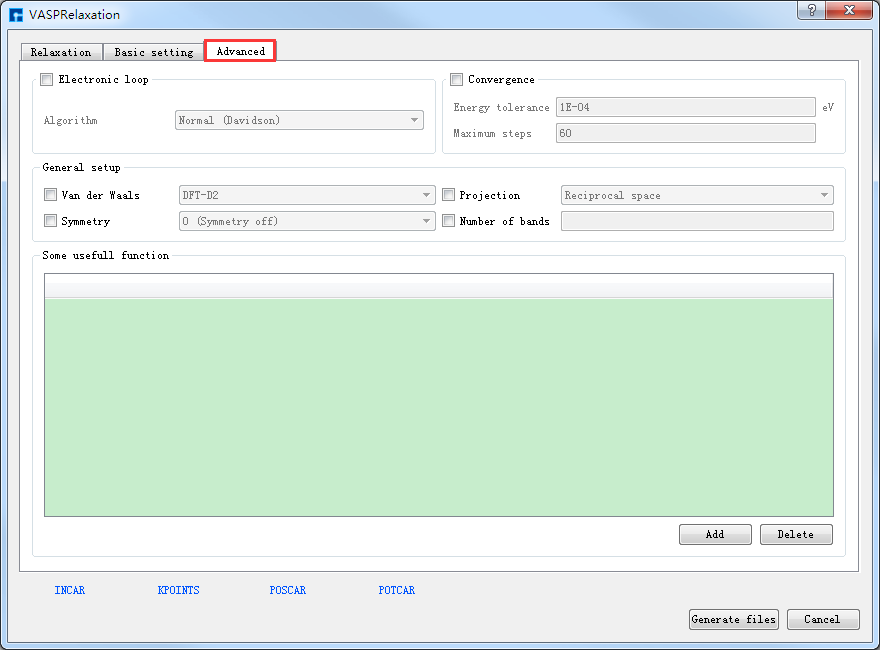
图 10.83 Advanced参数设置界面¶
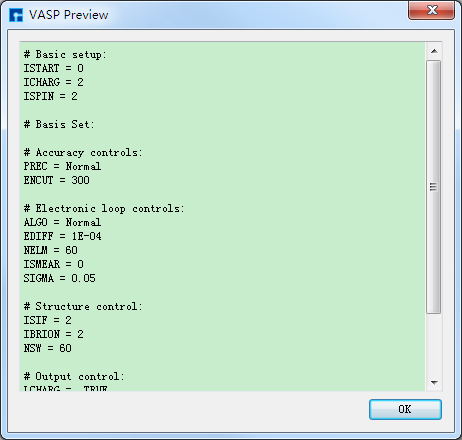
图 10.84 INCAR预览¶ |
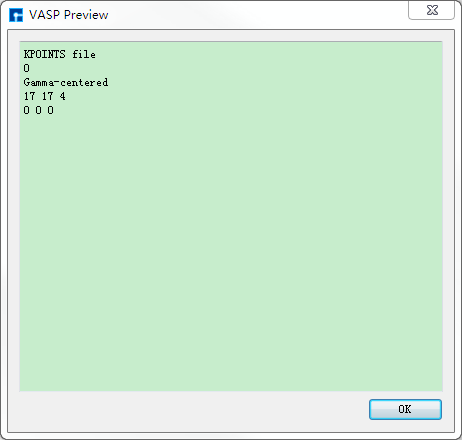
图 10.85 KPOINTS预览¶ |
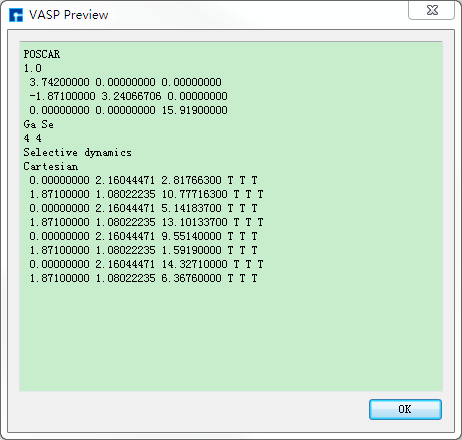
图 10.86 POSCAR预览¶ |
图 10.87 POTCAR预览¶ |
生成GaSe晶体结构的结构弛豫计算的输入文件 INCAR、KPOINTS、POSCAR、POTCAR 的Device Studio界面如 图 10.88 所示。
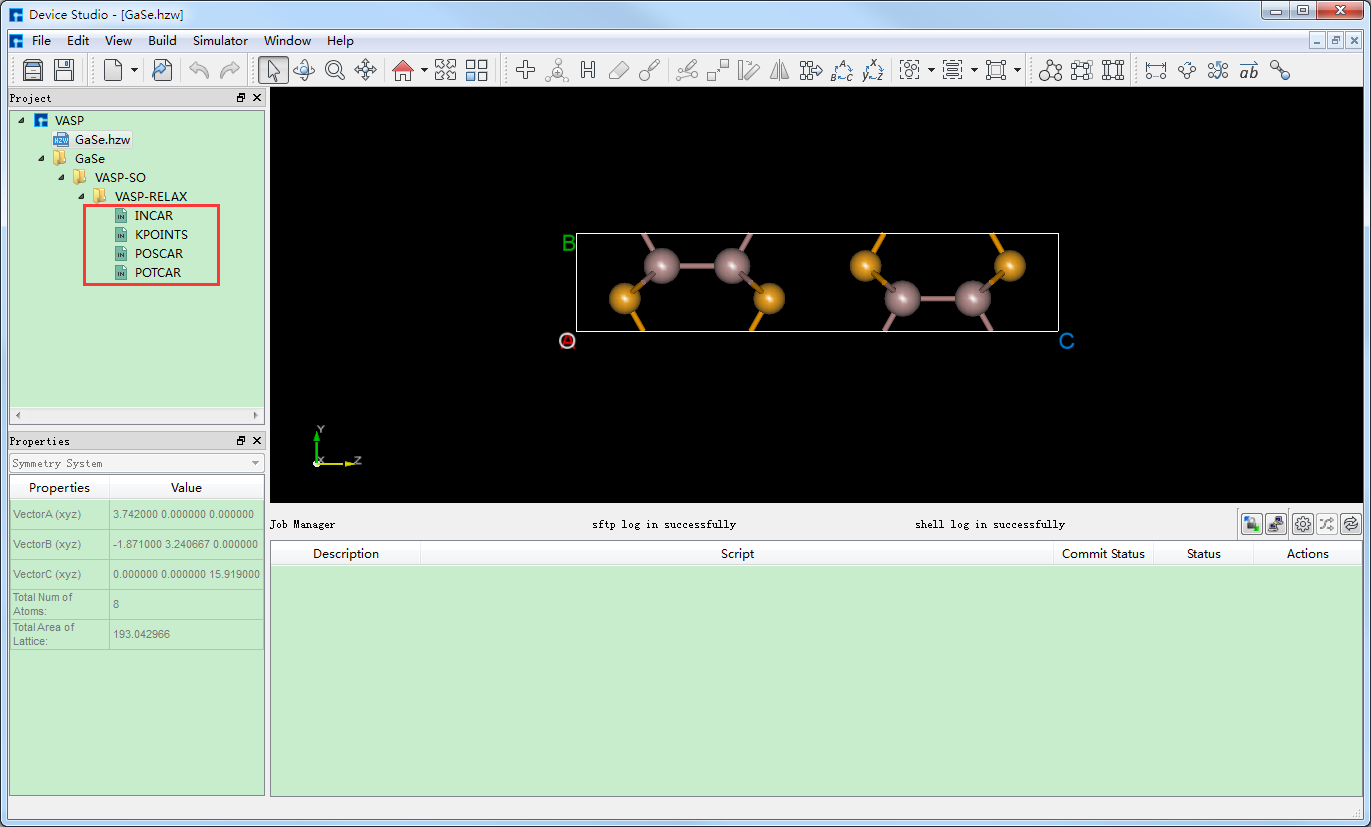
图 10.88 生成GaSe晶体结构的结构弛豫计算的输入文件的Device Studio图形界面¶
10.8.5. VASP计算¶
在做 GaSe晶体结构的结构弛豫计算 之前,需连接装有VASP的服务器,具体连接过程这里不做详细说明,用户可参考 Nanodcal连接服务器
节内容。连接好装有VASP的服务器,在做计算之前,用户可根据需要打开输入文件并查看文件中的参数设置是否合理,若不合理,则可选择直接在文件中编辑或重新生成,最后再进
行VASP计算(这里VASP计算指 GaSe晶体结构的结构弛豫计算)。如打开 INCAR 文件,在如 图 10.88 所示的界面中,选中 INCAR → 右击 →
Open with 即可查看到 INCAR 文件如下所示。对于其他输入文件,用户可根据需要选择是否打开查看,这里不做详细说明。
# Basic setup:
ISTART = 0
ICHARG = 2
ISPIN = 2
# Basis Set:
# Accuracy controls:
PREC = Normal
ENCUT = 300
# Electronic loop controls:
ALGO = Normal
EDIFF = 1E-04
NELM = 60
ISMEAR = 0
SIGMA = 0.05
# Structure control:
ISIF = 2
IBRION = 2
NSW = 60
# Output control:
LCHARG = .TRUE.
LWAVE = .FALSE.
LELF = .FALSE.
LVTOT = .FALSE.
LVHAR = .FALSE.
备注
在实际计算过程中,用户可通过 VASP 使用教程了解各参数的详细意义,从而根据计算需要合理设置参数。
在如 图 10.88 所示的界面中,选中 VASP-RELAX → 右击 → Run,弹出Run界面,在Run界面中点击 Save 按钮保存相应的脚本,再点击Run界面中的 Run 按钮则可做VASP计算。用户可在Job Manager区域中观察VASP计算状态,当VASP计算任务处于排队中、 计算中和计算完成时,Status 分别为Queued、Running、Finished。计算完成后,点击Job Manager区域 Action 下的 下载 按钮弹出下载VASP计算结果的界面,在该界面中点击 Download 则可下载,下载后可在软件的Project Explorer区域查看到该结果文件,如结果文件CHGCAR, 下载后可在Device Studio的Project Explorer区域看到该文件。
10.8.6. VASP计算结果的可视化分析¶
在Device Studio图形界面的Project Explorer区域内,选中结果文件 CHGCAR → 右击 → Show View,则弹出VASP计算结果电荷密度的可视化分析界面。
10.9. TOPS实例¶
Topological Polymeric Self-Consistent Field Theory 是一款基于自洽平均场理论(Self-Consistent Field Theory)的嵌段共聚物自组装相行为计算与模拟软件,简称 TOPS 。嵌段共聚物作为一种特殊的高分子,因其独特的分相机制,适宜的 分相尺度(10-100 nm)和在材料、半导体纳米刻蚀领域内的潜在应用受到人们的广泛关注和研究。但由于嵌段共聚物自组装体系参数空 间巨大且形成的自由能曲面非常复杂,其热力学平衡态结构的预测对于理论计算来说非常具有挑战性。
复旦大学李卫华教授课题组 结合其在嵌段共聚物自洽场计算领域内数十年的经验开发了这款软件TOPS,其具有以下功 能和特点:
精确计算几乎任意拓扑结构体系中的目标相结构的自由能,实现稳定相结构的预测、构建相图和分析机理。
适用于嵌段共聚物熔体体系、共混体系和溶液体系,为计算理论探讨和实验先行筛选出合适的参数空间。
拥有非常丰富的一元和二元结构库。
结合自动优化周期及特殊初始化算法,兼备高效率和高精度。
结合快速傅里叶变换(FFT)和Anderson Mixing,进一步加速了收敛过程。
可快速创建分子模型,可视化其结构和计算结果。
支持Windows和Linux平台,搭载鸿之微科技(上海)股份有限公司的Device Studio,可快速在本地和服务器递交任务并进行计算。
对石油化工、塑料加工、特种塑料盒纤维生产都有潜在的应用。
TOPS具有以下应用 :
构建相图 :TOPS 可以计算给定参数下备选结构的自由能,然后通过自由能的比较构建热力学平衡态结构的相图。
分析自由能数据 :TOPS 计算可以得到所有备选结构的自由能、界面能和熵。通过这些数据可以分析结构的稳定性几相关机制。
分析嵌段在结构中的分布 :TOPS 计算可以获得每个嵌段的密度分布,从而可以进一步探讨嵌段共聚物的自组装机理。
TOPS采用随机初始化,可以进行嵌段共聚物自组装形成新结构的探索,为实验制备新的有序结构提供指导 。
以 AB两嵌段共聚物计算BCC结构 为例来详细描述 TOPS 在Device Studio中的应用。

10.9.2. TOPS创建项目¶
双击Device Studio图标快捷方式,登录并启动Device Studio,在创建或打开项目界面中(启动软件后选择创建或打开项目的图形界面),根据界面提示选择创建一个新的项目( Create a new Project )或打开一个已经存在的项目( Open an existing Project )的按钮,选中之后点击界面中的 OK 按钮即可。若选择创建一个新的项目,用户可根据需要给该项目命名,如本项目命名为 TOPS,或采用软件默认项目名。
10.9.3. TOPS建模(分子结构)¶
以构建 AB两嵌段共聚物 为例,在Device Studio的图形界面中,选中 Simulator → TOPS → TOPS,弹出TOPS界面如 图 10.90 所示。
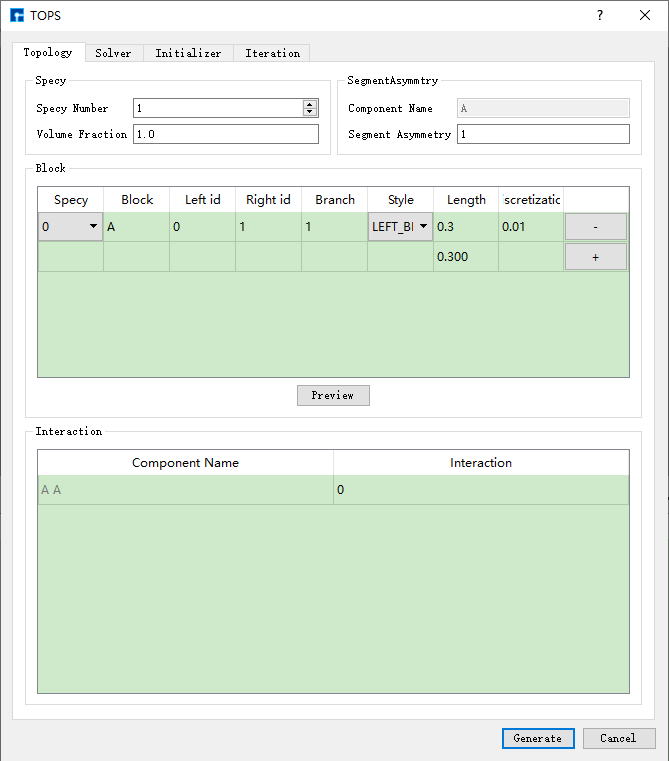
图 10.90 TOPS界面¶
在 图 10.90 所示界面中,设置参数如 图 10.91 中红色框选部分所示,点击 Preview 则可预览搭建好的 AB两嵌段共聚物 分子结构。
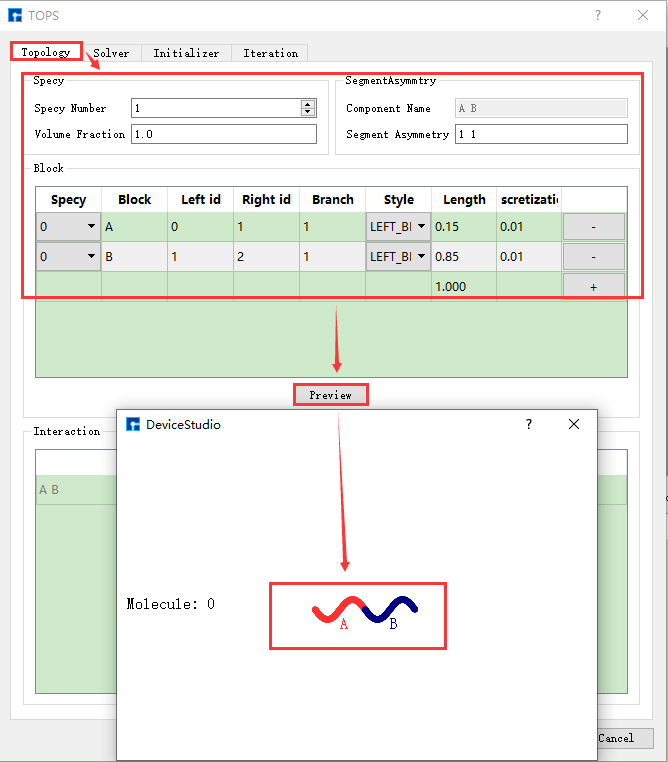
图 10.91 TOPS构建 AB两嵌段共聚物 界面¶
10.9.4. TOPS输入文件的生成¶
以生成 AB两嵌段共聚物计算BCC结构 的输入文件为例,在如 图 10.90 所示TOPS界面上分别选中 Topology、
Solver、Initializer、Iteration 设置参数分别如 图 10.92 、 图 10.93 、 图 10.94 和 图 10.95 所示,设置好参数后,点击TOPS界面中的 Generate 即可生成 AB两嵌段共聚物计算BCC结构 的输入文件 input 。
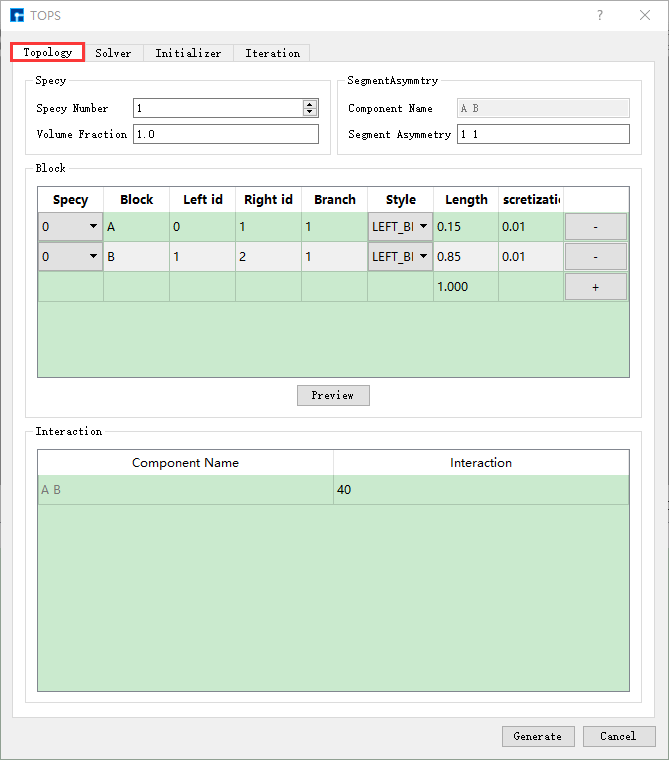
图 10.92 Topology参数设置界面¶
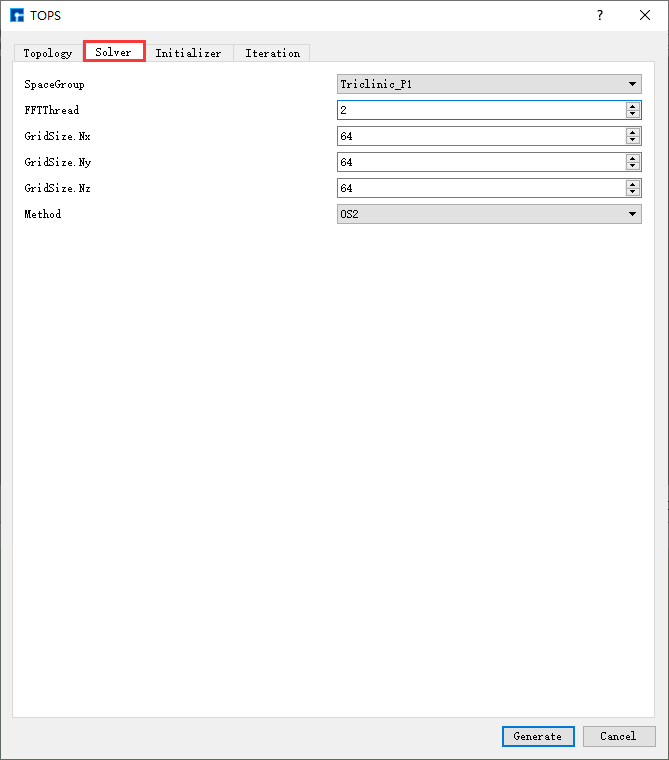
图 10.93 Solver参数设置界面¶
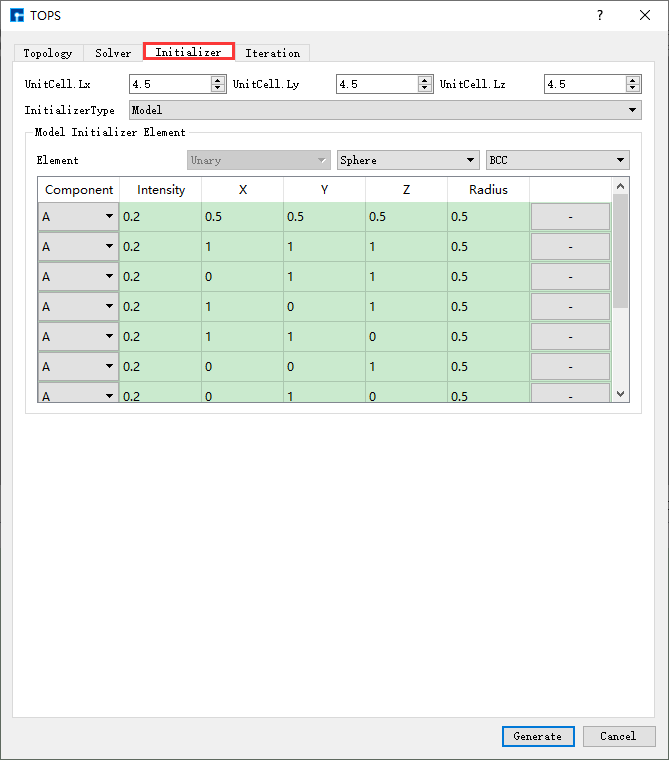
图 10.94 Initializer参数设置界面¶
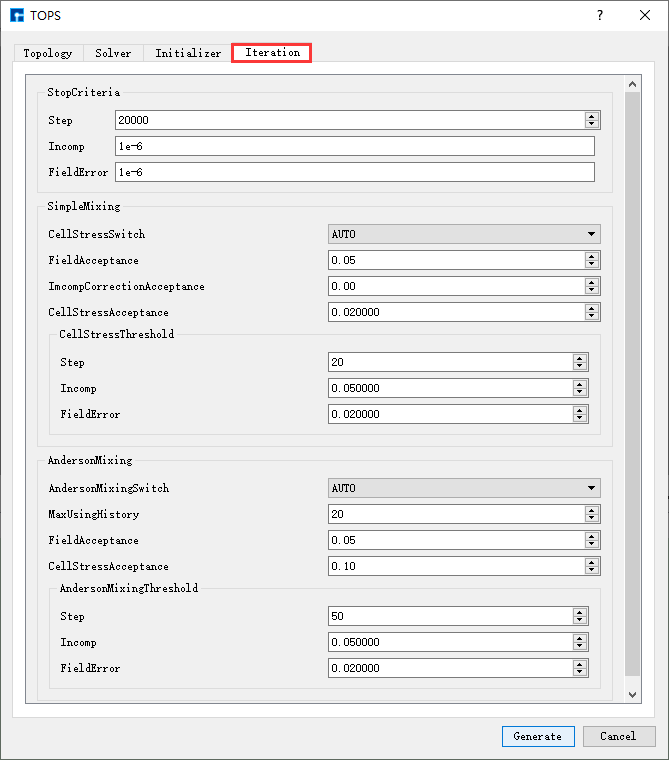
图 10.95 Iteration参数设置界面¶
生成 AB两嵌段共聚物计算BCC结构 输入文件 input 的Device Studio界面如 图 10.96 所示。
图 10.96 生成 AB两嵌段共聚物计算BCC结构 输入文件的Device Studio图形界面¶
10.9.5. TOPS计算¶
在做 AB两嵌段共聚物计算BCC结构 之前,需连接装有TOPS的本地电脑或服务器,具体连接过程这里不做详细说明,用户可参考 Nanodcal连接服务器
节内容。这里以在本地电脑上计算为例,连接好本地电脑后,在做计算之前,用户可根据需要打开输入文件查看文件中的参数设置是
否合理,若不合理,则可选择直接在文件中编辑或重新生成,最后再进行TOPS计算。如打开 input 文件,在8.9-5所示界面
中,选中 input → 右击 → Open with 即可查看到 input 文件如下所示。
Topology.Specy =
0 1.0
//SpecyID VolFrac
Topology.Block =
0 A 0 1 1 LEFT_BRANCH 0.15 0.01
0 B 1 2 1 LEFT_BRANCH 0.85 0.01
Topology.Interaction =
A B 40
Topology.SegmentAsymmtry =
A 1
B 1
Solver.SpaceGroup = Triclinic_P1
Solver.Method = OS2
Solver.FFTThread = 2
Solver.GridSize.Nx = 64
Solver.GridSize.Ny = 64
Solver.GridSize.Nz = 64
Initializer.UnitCell.Lx = 4.5
Initializer.UnitCell.Ly = 4.5
Initializer.UnitCell.Lz = 4.5
Initializer.InitializerType = Model
Initializer.ModelInitializer.Element =
Sphere A 0.2 0.5 0.5 0.5 0.5
Sphere A 0.2 1 1 1 0.5
Sphere A 0.2 0 1 1 0.5
Sphere A 0.2 1 0 1 0.5
Sphere A 0.2 1 1 0 0.5
Sphere A 0.2 0 0 1 0.5
Sphere A 0.2 0 1 0 0.5
Sphere A 0.2 1 0 0 0.5
Sphere A 0.2 0 0 0 0.5
Iteration.StopCriteria.Step = 20000
Iteration.StopCriteria.Incomp = 1e-6
Iteration.StopCriteria.FieldError = 1e-6
Iteration.SimpleMixing.FieldAcceptance = 0.05
Iteration.SimpleMixing.ImcompCorrectionAcceptance = 0
Iteration.SimpleMixing.CellStressSwitch = AUTO
Iteration.SimpleMixing.CellStressAcceptance = 0.02
Iteration.SimpleMixing.CellStressThreshold.Step = 20
Iteration.SimpleMixing.CellStressThreshold.Incomp = 0.05
Iteration.SimpleMixing.CellStressThreshold.FieldError = 0.02
Iteration.AndersonMixing.MaxUsingHistory = 20
Iteration.AndersonMixing.FieldAcceptance = 0.05
Iteration.AndersonMixing.AndersonMixingSwitch = AUTO
Iteration.AndersonMixing.CellStressAcceptance = 0.1
Iteration.AndersonMixing.AndersonMixingThreshold.Step = 50
Iteration.AndersonMixing.AndersonMixingThreshold.Incomp = 0.05
Iteration.AndersonMixing.AndersonMixingThreshold.FieldError = 0.02
在如 图 10.96 所示界面中,在Device Studio的Project Explorer区域选中 input → 右击 → Run,弹出Run界面,在Run界面中点击 Run 按钮则可做TOPS计算。用户可在Job Manager区域中观察TOPS计算状态,当TOPS计算任务处于排队中、
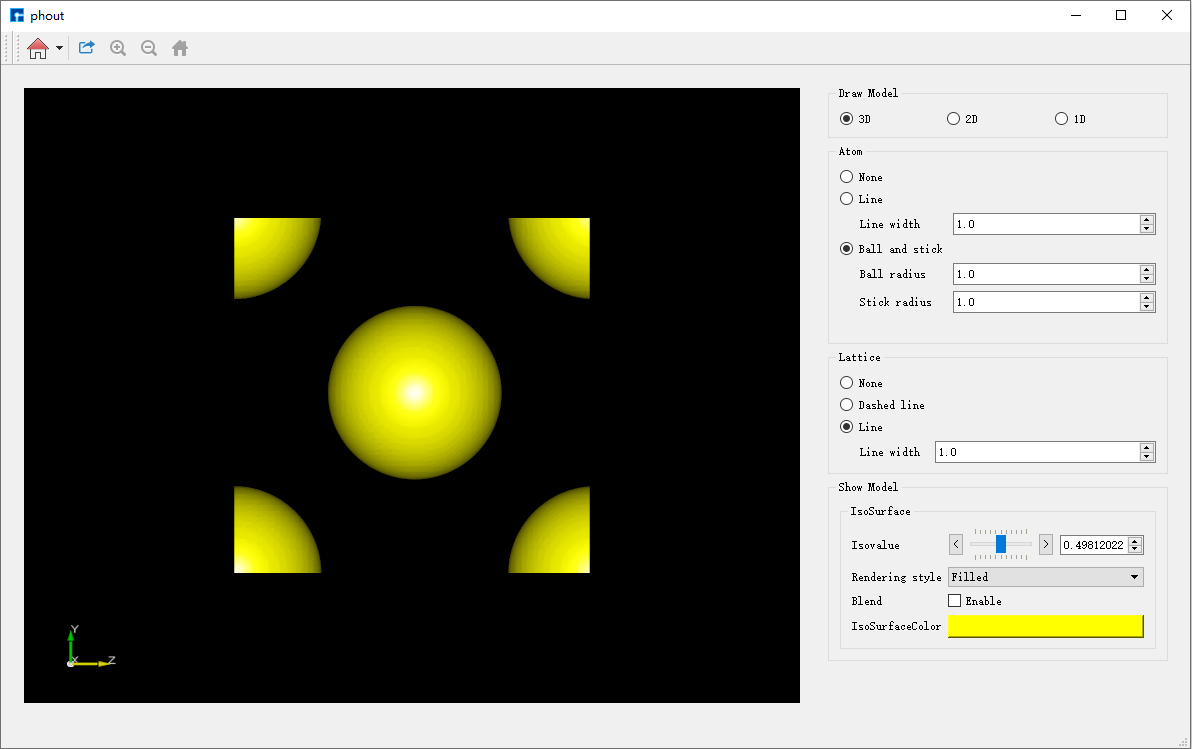
计算中和计算完成时,Status 分别为Queued、Running、Finished。计算完成后,选中 input → 右击 → Open Containing Folder 可看到TOPS计算完成的结果文件(密度文件) phout.txt 和记录文件(能量文件) printout.txt ,并找到其在本地电脑中存储位置。
10.10. DS-PAW 实例¶
DS-PAW 是Device Studio平台下的一款第一性原理平面波计算软件,使用平面波作为基函数组,赝势是使用投影缀加平面波方法构造的。 DS-PAW 功能强大,能够应用于不同场景,例如金属、半导体、绝缘体、表面、磁性、非磁性、锂电等; 能够精确预测材料的电子分布;能够进行原子几何结构优化;能够广泛的应用于材料科学领域。DS-PAW 性能稳定,在Intel芯片及国产海光芯片下经过百万案例的内部测试,包括各项功能及并行效率。
如 图 10.98 所示,DS-PAW 2021B版本已实现了 结构弛豫、电子结构、力学性质、磁性计算、过渡态、杂化泛函、范德瓦尔斯修正、光学性质、自旋轨道耦合、声子计算、强关联计算、第一性原理分子动力学 等共计28个计算功能,更多的功能仍在持续增加中。
以 Si晶体结构杂化泛函能带计算 为例来详细描述 DS-PAW 在Device Studio中的应用。
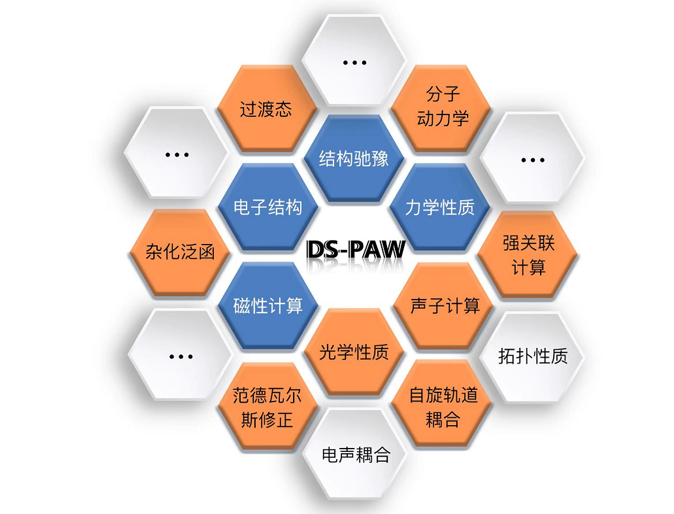
图 10.98 DS-PAW软件功能图¶

10.10.2. DS-PAW创建项目¶
双击Device Studio图标快捷方式,登录并启动Device Studio,在创建或打开项目界面中(启动软件后选择创建或打开项目的图形界面),根据界面提示选择创建一个新的项目( Create a new Project )或打开一个已经存在的项目( Open an existing Project )的按钮,选中之后点击界面中的 OK 按钮即可。若选择创建一个新的项目,用户可根据需要给该项目命名,如本项目命名为 DS-PAW,或采用软件默认项目名。
10.10.3. DS-PAW导入结构¶
在Device Studio的图形界面中点击 File → Import → Import Local,
则弹出导入DS-PAW结构文件的界面,根据界面提示找到 Si.hzw 结构文件的位置,选中 Si.hzw 结构
文件,点击 打开 按钮则导入 Si.hzw 结构后的Device Studio界面如 图 10.100 所示。在Device Studio中导入结构的其他方法这里不做详细说明,用户可参照 导入结构 节内容。
图 10.100 导入 Si.hzw 结构后的Device Studio图形界面¶
10.10.4. DS-PAW输入文件的生成¶
在如 图 10.100 所示界面中选中 Simulator → DS-PAW → Electronic Structure,弹出DS-PAW的参数设置界面Electronic structure如 图 10.101 所示。Electronic structure界面主要分为 Task 、 Solver setting 、 Physical setting 和 Advanced 四个模块,用户可根据计算需要,依次点击四个模块进行参数设置,之后点击 Generate files 即可生成对应计算的输入文件。
图 10.101 DS-PAW的参数设置界面Electronic structure¶
以生成 Si晶体结构杂化泛函能带计算 的输入文件为例,在如 图 10.101 所示的Electronic structure界面中,根据计算需要分别选
中 Task 、 Solver setting 、 Physical setting 和 Advanced ,设置参数
分别如图 图 10.102 、 图 10.103 、 图 10.104 和 图 10.105 所示,之后点击界面中的 Generate files 即可生成输入文件 scf.in 和 structure。
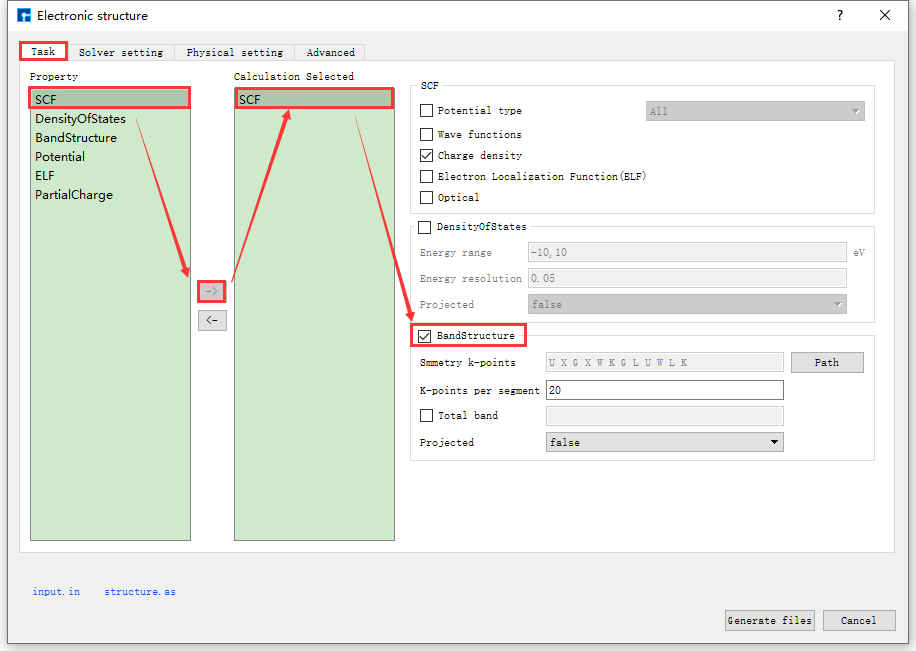
图 10.102 Task参数设置界面¶
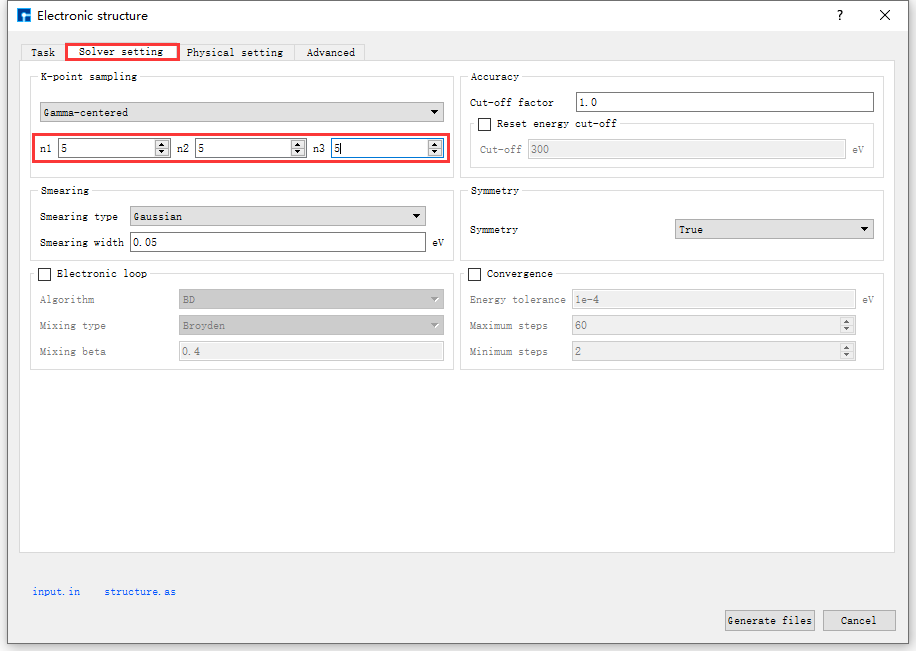
图 10.103 Solver setting参数设置界面¶
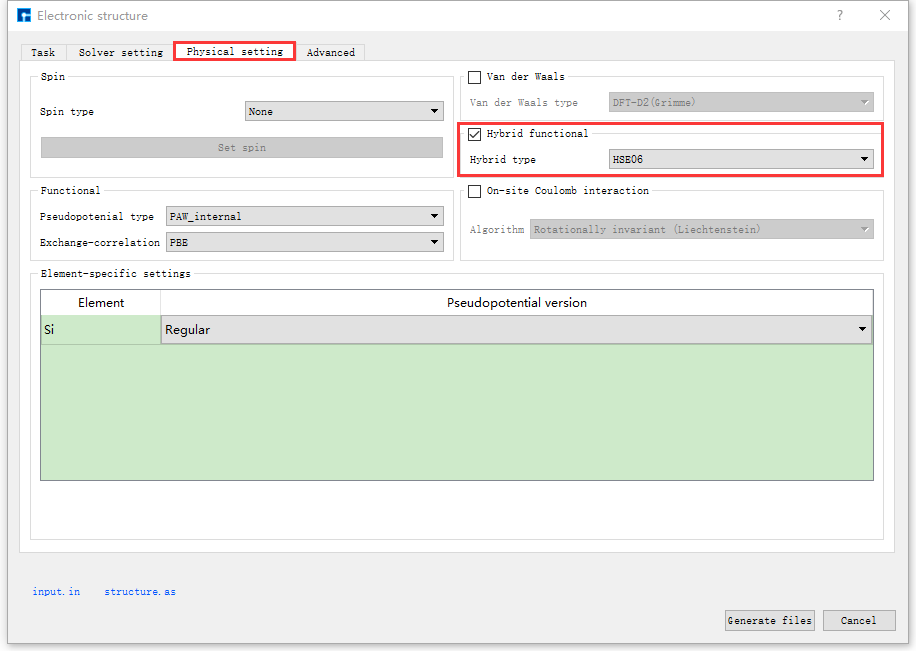
图 10.104 Physical setting参数设置界面¶
图 10.105 Advanced参数设置界面¶
生成 Si晶体结构杂化泛函能带计算 的输入文件 scf.in 和 structure 的Device Studio界面如 图 10.106 所示。
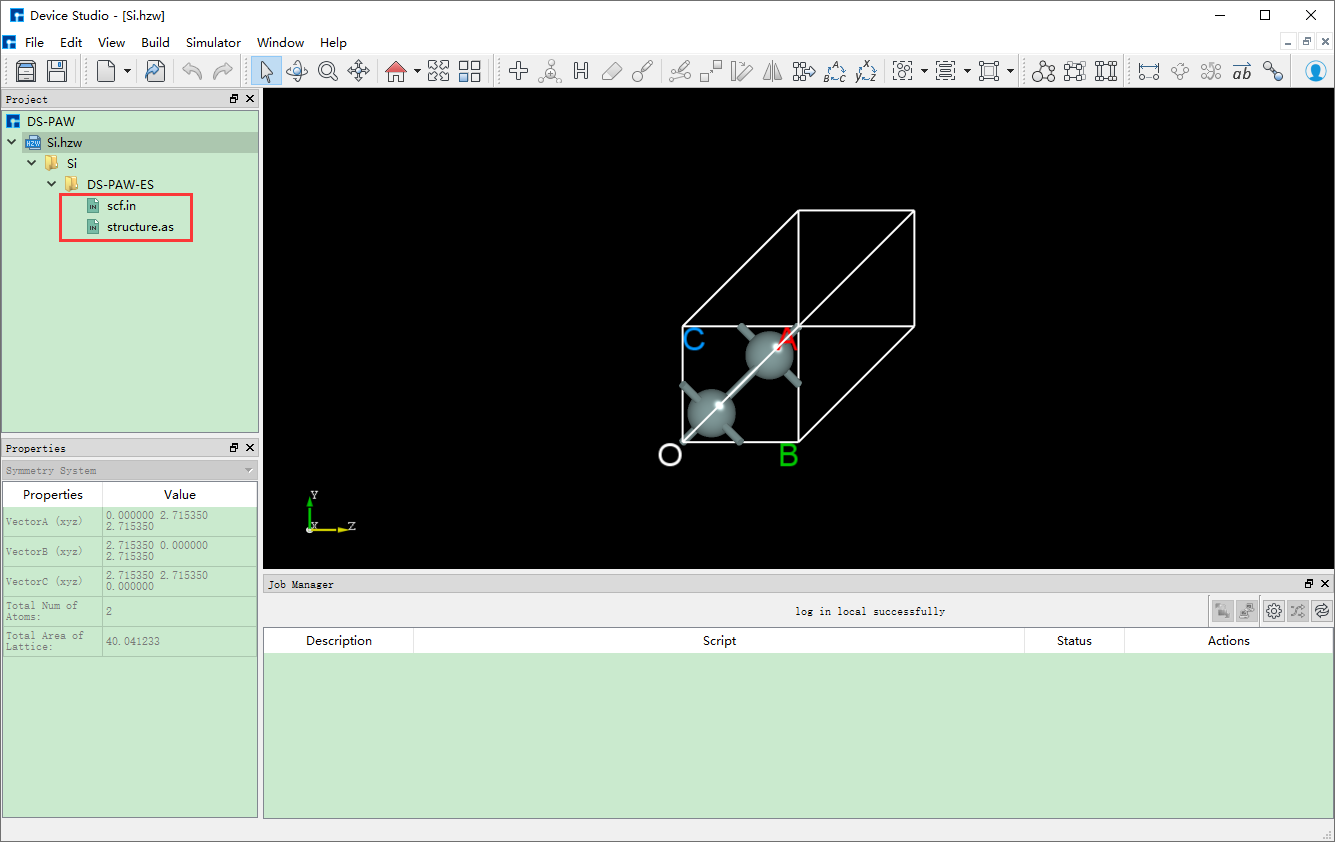
图 10.106 生成 Si晶体结构杂化泛函能带计算 输入文件的Device Studio界面¶
10.10.5. DS-PAW计算¶
在做 Si晶体结构杂化泛函能带计算 之前,需连接装有DS-PAW的本地电脑或服务器,具体连接过程这里不做详细说明,用户可参
考 Nanodcal连接服务器 节内容。这里以在本地电脑上计算为例,连接好装有DS-PAW的本地电脑后,在做计算之前,用户可根据需要
打开输入文件并查看文件中的参数设置是否合理,若不合理,则可选择直接在文件中编辑或重新生成,最后再进行DS-PAW计算。如打
开 scf.in 文件,在Device Studio的Project Explorer区域选中 scf.in → 右击 → Open with 即可查看到 scf.in 文件如 图 10.107 所示。对于其他输入文件,用户可根据需要选择是否打开查看,这里不做详细说明。
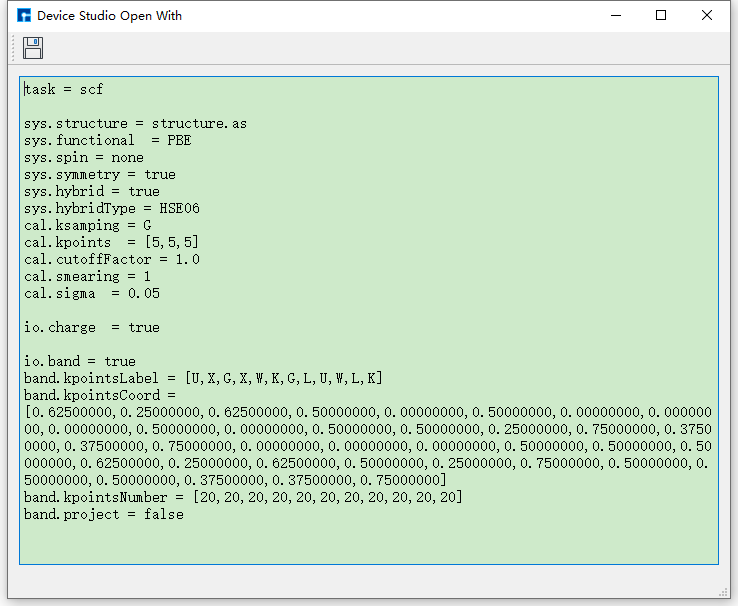
图 10.107 scf.in 文件¶
在 图 10.106 所示界面中,在Device Studio的Project Explorer区域选中 scf.in → 右击 → Run,弹出Run界面,在Run
界面中点击 Run 按钮则可做DS-PAW计算。用户可在Job Manager区域中观察DS-PAW的计算状态,当DS-PAW计算任务处于排队中、
计算中和计算完成时,Status 分别为Queued、Running、Finished。计算完成后,可在Device Studio的Project Explorer区域
中看到结果文件 band.json 和日志文件 DS-PAW.log。
10.10.6. DS-PAW计算结果的可视化分析¶
在Device Studio的Project Explorer区域中选中 band.json → 右击 → Show View,则弹出DS-PAW计算结果的可视化分
析界面如 图 10.108 所示。
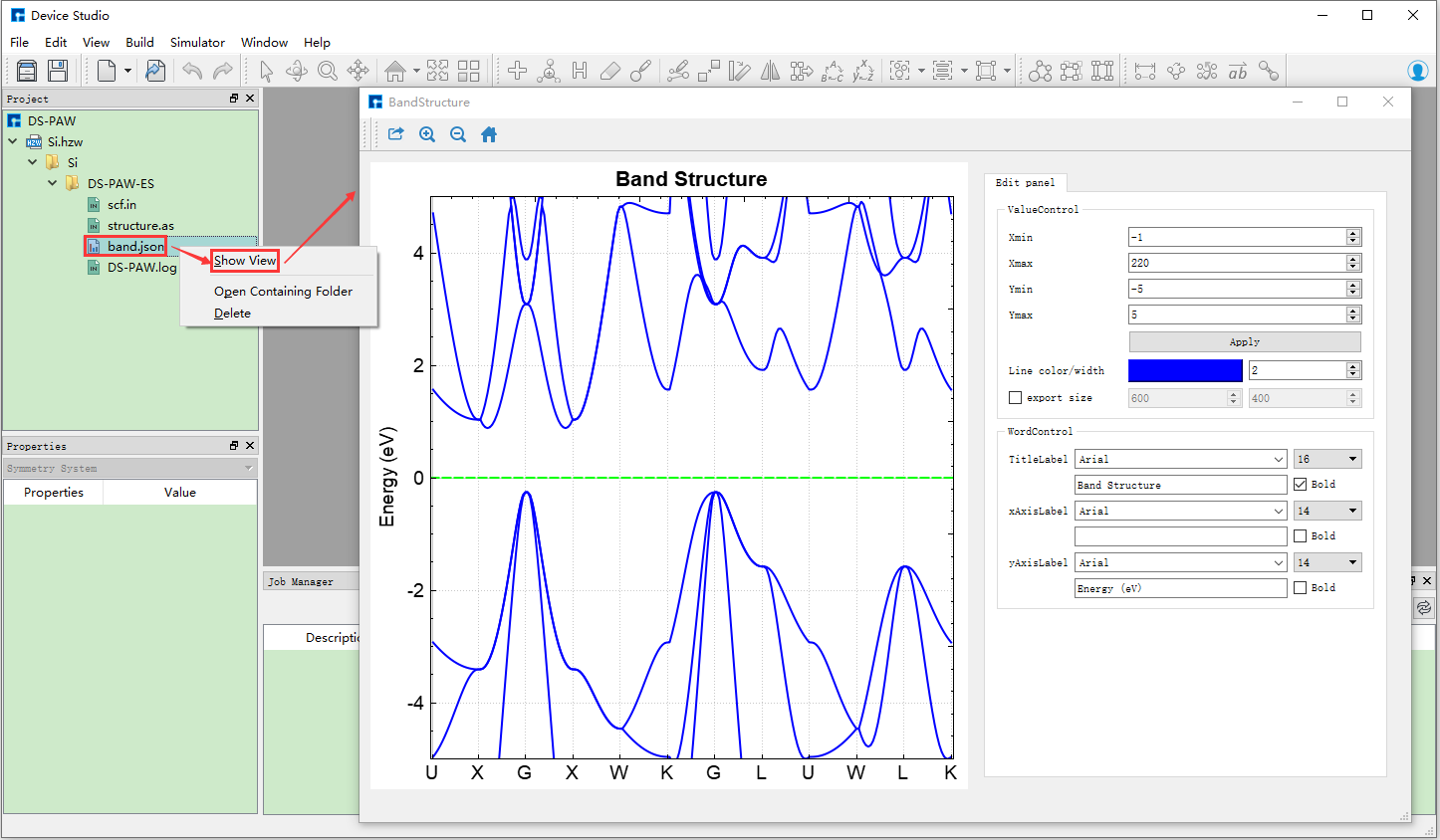
图 10.108 DS-PAW计算结果的可视化分析界面¶
注意事项
DS-PAW的计算结果往往以 .json 或 .h5 格式存储,目前Device Studio支持DS-PAW小于 68MB 的 .json 格式的结果文件的可视化分析。对于大于 68MB 的 .json 文件建议用户使用DS-PAW辅助工具包 dspawpy 进行数据分析。
10.11. BDF 实例¶
BDF (Beijing Density Functional)是一个独立完整、具有完全自主知识产权的 量子化学计算软件包。也是国际上第一个 基于现代密度泛函理论、能准确计算分子体系基态总能量的完全相对论密度泛函程序 (早期的类似程序因数值积分精度太差不能准确 计算总能量)。
BDF 官方用户手册网址 为:https://bdf-manual.readthedocs.io/zh_CN/latest/index.html
BDF的研发团队信息:
国际量子分子科学院院士刘文剑研发团队.
鸿之微研发团队、服务团队。
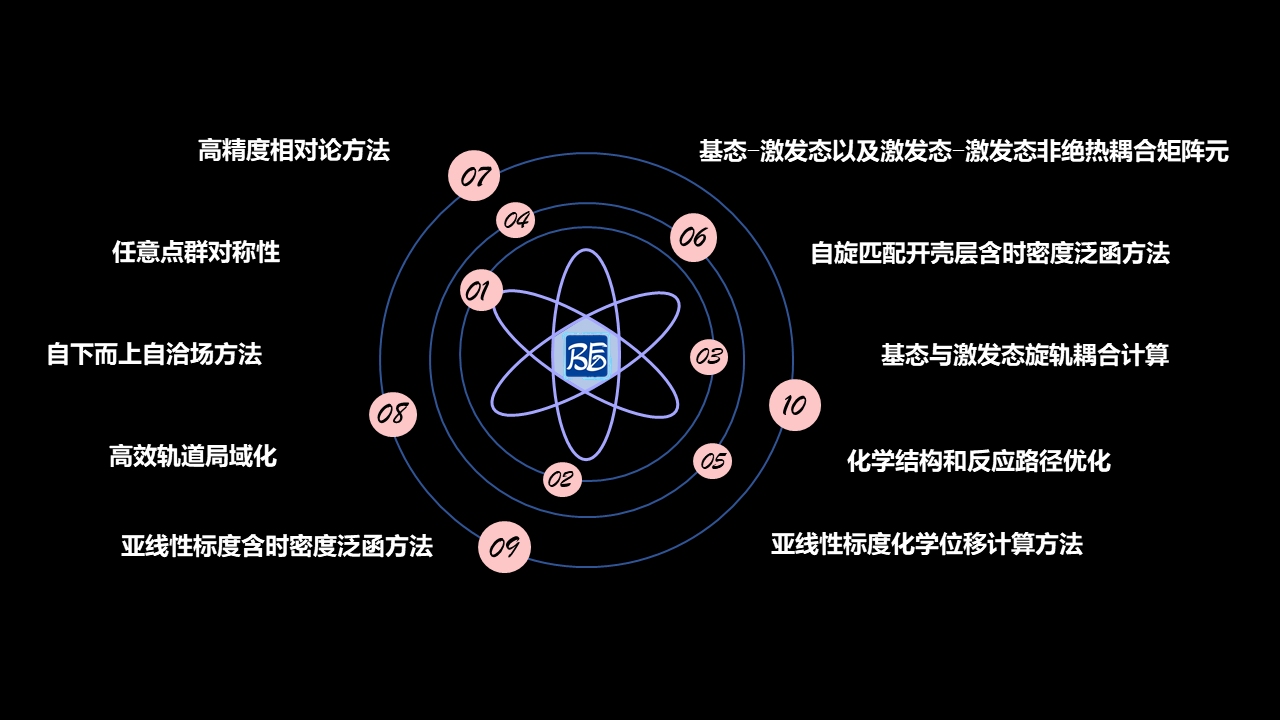
图 10.109 BDF软件特色图¶
BDF软件特色:
高精度相对论方法。
任意点群对称性。
自下而上自洽场方法。
高效轨道局域化。
亚线性标度含时密度泛函方法。
亚线性标度化学位移计算方法。
自旋匹配开壳层含时密度泛函方法。
基态与激发态旋轨耦合计算。
基态——激发态以及激发态——激发态非绝热耦合矩阵元。
化学结构和反应路径优化。
以 基态分子CH2S完成sf-X2C/TDDFT-SOC的自旋轨道耦合计算 为例来详细描述 BDF 在Device Studio中的应用。
10.11.1. BDF的发展史¶
BDF 的研发始于1993年,并于1997年正式命名。最初的想法是 对稀土、锕系、过渡金属、超重元素等小分子体系进行高精度计算,考察这些体系中的相对论效应,因此一开始就采用基于Dirac算符的完全相对论密度泛函理论(4C-DFT)和近乎完备的基函数“数值基+STO”(Slater-type orbital)。正因为如此,BDF对稀土、锕系、超重元素的计算结果一直被作为检验其他近似相对论方法的基准。BDF对重元素体系电子、分子结构的计算结果被后续20余个实验验证。
2009年开始引入 基于高斯基的解析积分,BDF 进入一个新的发展阶段。
毋庸讳言,BDF 一开始就定位于发展新理论、新方法和新算法的平台,因此是一款“科研软件”。基于 BDF 发展的理论和方法包括:相对论含时密度泛函理论(4C/ZORA/X2C-TDDFT)、精确二分量(X2C)相对论理论、准四分量(Q4C)相对论理论、自旋分离的X2C相对论理论(sf-X2C+so-DKHn)、多体有效量子电动力学(eQED)、相对论核磁理论(4C/X2C-NMR)、相对论核自旋-转动理论(4C-NSR)、相对论能带理论(X2C-PBC)、X2C解析梯度和Hessian;激发态HF/KS方法(mom);基于“用分子片合成分子”(F2M)思想的轨道局域化方案(FLMO)、亚线性标度含时密度泛函理论(FLMO-TDDFT)、亚线性标度NMR方法(FLMO-NMR)、迭代轨道相互作用“自下而上”自洽场方法(iOI);自旋匹配开壳层含时密度泛函理论(SA-TDDFT)、自旋反转含时密度泛函理论(SF-TDDFT)、基态/激发态-激发态非绝热耦合含时密度泛函理论(NAC-TDDFT)、含时密度泛函理论解析能量梯度、任意单值/双值点群对称化等。
除了上述相对论/非相对论密度泛函、含时密度泛函理论,BDF 还有基于“先静态再动态又静态”(SDS)思想的波函数电子相关方法SDSPT2SDSCI、iCI、iCIPT2、iCAS、iCISCF、SOC-iCI、iCI-SOC,以及直接求解大矩阵内部本征态的iVI方法等等。
鉴于 BDF 的现状,首期商业化版本将以荧/磷光材料发光机理和材料设计为主要应用目标,因此不包括4C/X2C相对论、波函数电子相关、固体能带/核磁等方法。即首期商业化的BDF将以DFT、TDDFT为主,包括基态与激发态KS、QM/MM、FLMO-TDDFT、SF-TDDFT、NAC-TDDFT、sf-X2C-SA-TDDFT/SOC、SA-TDDFT解析能量梯度和数值Hessian、稳定结构和过渡态优化、反应路径优化、隐式溶剂化模型、FLMO-NMR、基于局域轨道(FLMO)的性质计算和分析等特色功能。

10.11.3. BDF创建项目¶
双击Device Studio图标快捷方式,登录并启动Device Studio,在创建或打开项目界面中(启动软件后选择创建或打开项目的图形界面),根据界面提示选择创建一个新的项目( Create a new Project )或打开一个已经存在的项目( Open an existing Project )的按钮,选中之后点击界面中的 OK 按钮即可。若选择创建一个新的项目,用户可根据需要给该项目命名,如本项目命名为 BDF,或采用软件默认项目名。
10.11.4. BDF导入结构¶
在Device Studio的图形界面中点击 File → Import → Import Local,
则弹出导入BDF结构文件的界面,根据界面提示找到 CH2S.hzw 结构文件的位置,选中 CH2S.hzw 结构
文件,点击 打开 按钮则导入 CH2S.hzw 结构后的Device Studio界面如 图 10.111 所示。在Device Studio中导入结构的其他方法这里不做详细说明,用户可参照 导入结构 节内容。
图 10.111 导入 CH2S.hzw 结构后的Device Studio图形界面¶
10.11.5. BDF输入文件的生成¶
在如 图 10.111 所示界面中选中 Simulator → BDF → BDF,弹出BDF的参数设置界面BDF Job Setup如 图 10.112 所示。
以生成 基态分子CH2S完成sf-X2C/TDDFT-SOC的自旋轨道耦合计算 的输入文件为例,在如 图 10.111 所示的BDF Job Setup界面中,根据计算需要分别选中 Basic Settings 、 SCF Settings 、 TDDFT Settings 和 Preview Script ,设置参数
分别如 图 10.112 、 图 10.113 、 图 10.114 和 图 10.115 所示,之后点击界面中的 Generate files 即可生成输入文件 bdf.inp。
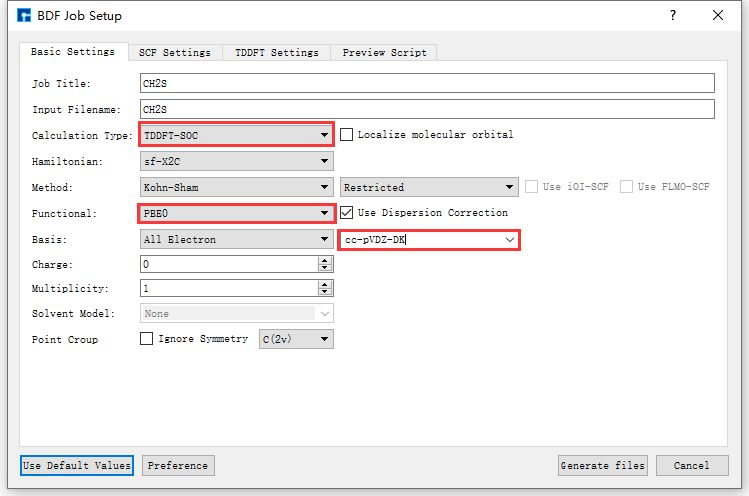
图 10.112 Basic Settings参数设置界面¶
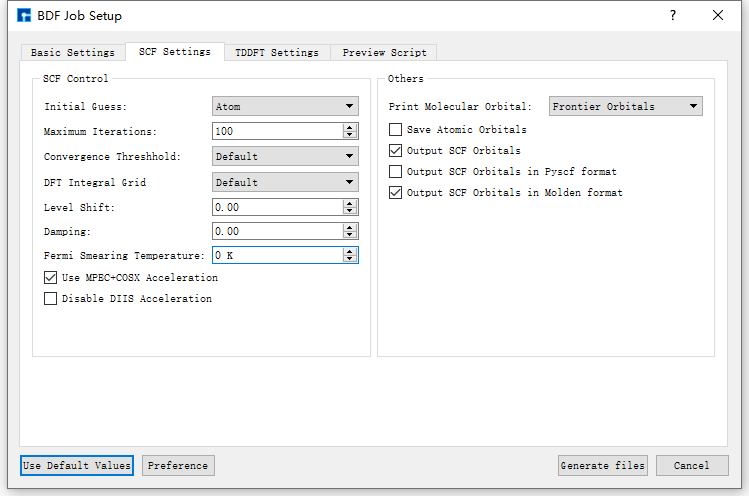
图 10.113 SCF Settings参数设置界面¶
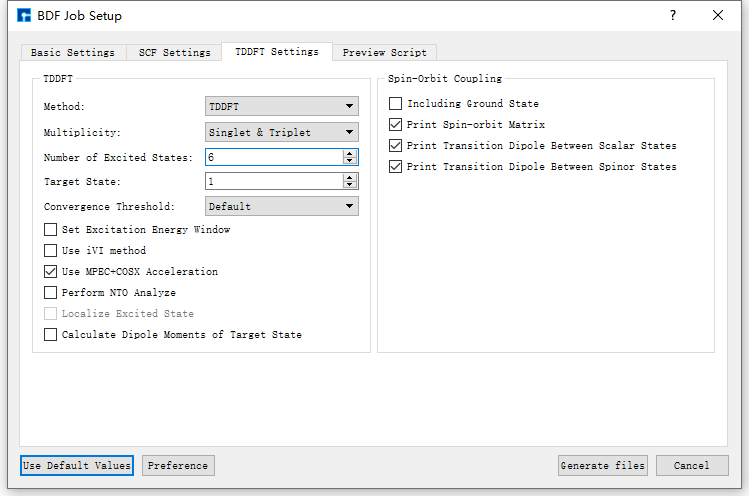
图 10.114 TDDFT Settings参数设置界面¶
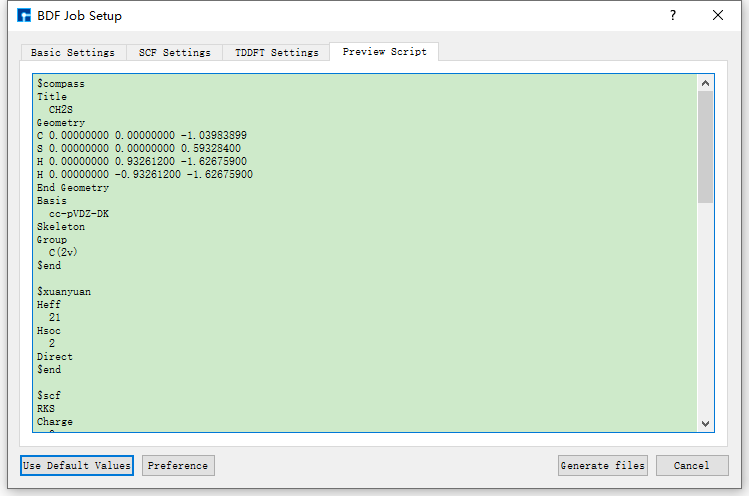
图 10.115 Preview Script参数设置界面¶
生成 基态分子CH2S完成sf-X2C/TDDFT-SOC的自旋轨道耦合计算 的输入文件 bdf.inp 的Device Studio界面如 图 10.116 所示。
图 10.116 生成 基态分子CH2S完成sf-X2C/TDDFT-SOC的自旋轨道耦合计算 输入文件的Device Studio界面¶
10.11.6. BDF计算¶
在做 基态分子CH2S完成sf-X2C/TDDFT-SOC的自旋轨道耦合计算 之前,需连接装有BDF的服务器,具体连接过程这里不做详细说明,用户可参
考 Nanodcal连接服务器 节内容。连接好装有BDF的服务器,在做计算之前,用户可根据需要打开输入文件并查看文件中的参数设置是否合理,若不合理,则可选择直接在文件中编辑或重新生成,最后再进行BDF计算(这里BDF计算指 基态分子CH2S完成sf-X2C/TDDFT-SOC的自旋轨道耦合计算 )。如打开 bdf.inp 文件,在Device Studio的Project Explorer区域选中 bdf.inp → 右击 → Open with 即可查看到 bdf.inp 文件如下所示。
$compass
Title
CH2S
Geometry
C 0.00000000 0.00000000 -1.03983899
S 0.00000000 0.00000000 0.59328400
H 0.00000000 0.93261200 -1.62675900
H 0.00000000 -0.93261200 -1.62675900
End Geometry
Basis
cc-pVDZ-DK
Skeleton
Group
C(2v)
$end
$xuanyuan
Heff
21
Hsoc
2
Direct
$end
$scf
RKS
Charge
0
SpinMulti
1
DFT
PBE0
D3
MPEC+COSX
Molden
$end
$tddft
Imethod
1
Isf
0
Idiag
1
Iroot
6
MPEC+COSX
Istore
1
$end
$tddft
Imethod
1
Isf
1
Idiag
1
Iroot
6
MPEC+COSX
Istore
2
$end
$tddft
Isoc
2
Nfiles
2
Imatsoc
-1
Imatrsf
-1
Imatrso
-1
$end
备注
在实际计算过程中,用户可通过 BDF 用户手册 了解各参数的详细意义,从而根据计算需要合理设置参数。若想详细了解 BDF ,点击对应紫色或蓝色字体软件名称,或发送邮件到邮箱 support@hzwtech.com 咨询。
在如图11-5所示界面中,在Device Studio的Project Explorer区域,选中 bdf.inp → 右击 → Run,弹出Run界面,在Run
界面中点击 Run 按钮则可做BDF计算。用户可在Job Manager区域中观察BDF的计算状态,当BDF计算任务处于排队中、
计算中和计算完成时,Status 分别为Queued、Running、Finished。计算完成后,点击Job Manager区域的Actions按钮,则弹出Qsftp Explorer界面,在该界面中,找到计算结果文件,点击 下载 按钮即可将计算结果文件从服务器下载到本地,下载后的结果文件可在Device Studio的Project Explorer区域查看到。