2 功能介绍¶
本章将依次介绍PASP的常用功能。对于每种功能,将简要介绍使用方法,并提供一组示例输入文件(为节省篇幅,这些文件另附在 example_public 文件夹中;在下文提到示例输入文件时,会指出具体路径)。初次使用某个功能时,建议先用示例输入文件测试运行一次,确认能够得到与手册内容一致的结果后,再换到需要研究的体系进行计算。在更换体系重新计算时,通常需要相应地修改若干设置,这时通常需要仔细研读示例输入文件中对各个设置项的注释,或者借助本手册第六章的“设置项索引”查看相应设置项的介绍。
2.1 简单磁性体系的有效哈密顿量方法¶
2.1.1 常用自旋哈密顿量模型¶
对于磁性体系而言,有效哈密顿量方法也称有效自旋哈密顿量方法。有效自旋哈密顿量最简单的形式为经典的海森堡模型(Heisenberg model,这里略去不重要的常数项 \(E_{0}\) ):
其中 \(S_{i}\) 与 \(S_{j}\) 表示原子 \(i\) 与 \(j\) 的总自旋矢量,近似视为经典矢量; \(e_{i}\) 与 \(e_{j}\) 表示 \(S_{i}\) 与 \(S_{j}\) 对应的单位方向向量。最后的等号后面是将各组等价的相互作用合并后的结果,这里假定只考虑前 \(m_{max}\) 种最近邻的不等价海森堡相互作用。
上述模型还可以推广为矩阵形式:
其中 \({ \mathbb{A}}_{i}\) 与 \({ \mathbb{J}}_{i j}\) ( \(\tilde { \mathbb{A}}_{i}\) 与 \(\tilde { \mathbb{J}}_{i j}\) )都是3×3矩阵,分别称为单离子各向异性(single-ion anisotropy, SIA)矩阵与J矩阵。J矩阵中包含了海森堡相互作用、Dzyaloshinskii-Moriya相互作用(DM相互作用)和Kitaev相互作用的信息。最后的等号后面是将各组等价的相互作用合并后的结果。公式 (2) 相比于公式 (1) 多出来的项是由自旋轨道耦合(SOC)导致的。
除此之外,有时还需考虑更高阶的相互作用。为简单起见,这里仅补充考虑四阶的点乘形式相互作用:
其中包含两体、三体和四体的贡献,这里分别用字母 \(B\) 、 \(T\) 、 \(Q\) 表示相应的参数。最后的等号后面是将各组等价的相互作用合并后的结果。
在不考虑SOC时,通常总的自旋哈密顿量可写成:
而在考虑SOC时,总的自旋哈密顿量写成:
以上两式中的 \(E_{0}\) 为常数项。由于能量零点的选择并不影响物理性质,通常 \(E_{0}\) 项不重要,可以省略。
为了构造具体体系的有效自旋哈密顿量模型,需要设置适当的截断距离(对于不同类型的相互作用,可以设定不同的截断距离),分析截断距离以内的不等价的磁性原子对(pair)或磁性原子簇(cluster),然后相应地计算各个不等价的相互作用系数。
在相对简单的磁性体系中,我们可以采用向红军教授提出的四态法 9 10 (见2.1.2节介绍)计算 \(H_{2}\) 或 \(H_{2,SOC}\) 中涉及的所有二阶相互作用参数,另外还可用四态法计算二体四阶的点乘形式相互作用(即双二次相互作用)参数 \(\left\{{B_{m^{\prime}}}\right\}\) 。对于更复杂的体系,可能需要考虑更多类型的相互作用,这时需要更具通用性的方法,参见2.2节内容。
2.1.2 四态法¶
2.1.2.1 算法简介¶
要计算公式 (1) 中各向同性海森堡相互作用 \(J_{i j}\) ,需作四次“不考虑SOC”的第一性原理计算:其中 \(i\) 与 \(j\) 原子处自旋矢量分别指向“ \(\uparrow \uparrow\) ”、“ \(\uparrow \downarrow\) ”、“ \(\downarrow \uparrow\) ”、“ \(\downarrow \downarrow\) ”( \(\uparrow\) 和 \(\downarrow\) 分别指沿 \(z\) 轴正反方向),而其他原子自旋均任意并在四次计算中保持不变(建议沿 \(x\) 或 \(y\) 轴,以避免部分高阶相互作用干扰),则参数 \(J_{i j}\) 和 \(\tilde{J}_{i j}\) 可由下式计算:
式中 \(S\) 为自旋矢量的模长, \(E_{i j,\alpha \beta }\left (\alpha,\beta =\uparrow,\downarrow\right) \) 与 \(j\) 原子处自旋方向分别 \(\alpha\) 为 \(\beta\) 与时第一性原理计算所得体系的总能量。以下各参数计算公式仅以不带波浪号(tilde)的参数为例,带波浪号的参数计算式需要相应地除以 \(S^{2}\) 。
公式 (2) 的 \({ \mathbb{J}}_{i j}\) 与 \({ \mathbb{A}}_{i}\) 矩阵中任意一个非独立成分都可由四种指定自旋构型的总能量(由考虑SOC的第一性原理计算所得)解出。对于 \({ \mathbb{J}}_{ij,ab}\left ( a,b=x,y,z \right )\) ,用 \(E_{i j,ab,\alpha \beta }\left (\alpha,\beta =\uparrow,\downarrow\right)\) 表示如下构型的第一性原理计算所得能量:原子 \(i\) 的自旋平行 \(\left ( \alpha=\uparrow \right )\) 或反平行 \(\left ( \alpha=\downarrow \right )\) 于 \(a\) 方向、原子 \(j\) 的自旋平行 \(\left ( \beta=\uparrow \right )\) 或反平行 \(\left ( \beta=\downarrow \right )\) 于 \(b\) 方向、其他原子自旋采用沿 \(c\) (指平行或反平行)方向 \(\left (c=x,y,z;c\ne a;c\ne b\right ) \) 可表达为
对于 \({ \mathbb{A}}_{i}\) 的非对角元 \({\mathbb{A}}_{i,ab}\left (a,b=x,y,z; a\ne b\right )\) ,用 \(E_{i,ab,\alpha \beta }\left (\alpha,\beta =\uparrow,\downarrow\right)\) 表示如下构型的第一性原理计算所得能量:原子 \(i\) 的自旋方向矢量的 \(\alpha\) 分量为 \(\pm \sqrt{2}/2\) (正负号对应于 \(\alpha =\uparrow,\downarrow\) ), \(b\) 分量为 \(\pm \sqrt{2}/2\) (正负号对应于 \(\beta =\uparrow,\downarrow\) ),剩下的 \(c\) 分量为 \(0=\left ( c=x,y,z;c\ne a;c\ne b \right )\) ;其余原子采用沿 \(c\) 方向的适当的构型并在四次计算中保持不变。 则 \({ \mathbb{A}}_{i,ab}\) 可表达为
而对于 \({ \mathbb{A}}_{i}\) 的对角元之差 \(\left ({\mathbb{A}}_{i,aa}-{\mathbb{A}}_{i,bb}\right )\) \(\left (a,b=x,y,z;a\ne b\right )\) ,用 \({E}_{i,ab,\alpha \beta }\left (\alpha ,\beta =\uparrow,\downarrow,0 \right)\) 表示如下构型的第一性原理计算所得能量:原子 \(i\) 的自旋方向矢量的 \(a\) 分量为 \(\pm 1\) 或 \(0\) (对应于 \(\alpha =\uparrow,\downarrow ,0\) ), \(b\) 分量为 \(\pm 1\) 或 \(0\) (对应于 \(\beta=\uparrow,\downarrow ,0\) ),剩下的 \(c\) 分量为 \(0 \left (c=x,y,z;c\ne a;c\ne b \right)\) ;其余原子采用沿 \(c\) 方向的适当的构型并在四次计算中保持不变。则 \(\left (\mathbb{A}_{i,aa}-\mathbb{A}_{i,bb} \right)\) 可表达为
双二次相互作用 \(B_{ij}\left(e_{i}\cdot e_{j} \right)^{2}\) 的系数 \(B_{ij}\) 也可用四态法计算。令 \(S_{i}\) 总指向 \(\left(1,0,0 \right)\) 方向, \(S_{j}\) 依次指向 \(\left(1,0,0 \right)\) 、 \(\left(-1,0,0 \right)\) 、 \(\left(1/\sqrt{2},1/\sqrt{2},0 \right)\) 、 \(\left(-1/\sqrt{2},-1/\sqrt{2},0 \right)\) 方向,其他自旋采用沿 \(z\) 方向的适当的构型并在四次计算中保持不变。这四种自旋构型的“不考虑SOC”的第一性原理计算所得总能量依次记为 \(E_{1}\) 、 \(E_{2}\) 、 \(E_{3}\) 、 \(E_{4}\) ,则 \(B_{ij}\) 可表达为
2.1.2.2 找不等价代表对 (Model find_J_path_sym)¶
在四态法中,仅涉及单体和两体的参数。对于单体,我们仅需找出有几种环境不等价的磁性原子,每种类型仅需挑出一个原子作为代表,用四态法算出它的 \({ \mathbb{A}}_{i}\) ,而与之等价的各个原子的 \(\left \{{ \mathbb{A}}_{i} \right \}\) 都可通过对称性推出(在PASP软件内可以自动完成)。同理,对于两体,在指定的截断距离内,我们也需要找出有几种环境不等价的磁性原子对(pair),然后每种类型需要挑出一对原子作为代表,用四态法算出它们的 \({ \mathbb{J}}_{ij}\) (有时还需算出 \(B_{ij}\) ),而与之等价的原子对的相应参数也可通过对称性推出(在PASP软件内可以自动完成)。
单体的代表原子的选择容易通过观察确定,这里省略。对于两体的代表对,也可通过观察确定,但当考虑的近邻数较多时会有些困难,此时可以利用PASP的 “Model find_J_path_sym” 方便地找出各个不等价代表对。
这里以 \(BiFeO_{3}\) 为例,使用空间群为R3c的原胞,暂时先假定使用2×2×2超胞,所需的全部输入文件在 example_public/find_J_path_sym/BiFeO3_222cell/input 文件夹下,包括 unit_cell.str (原胞信息)、 cell.str (超胞信息)和 PASP.input (主要输入文件)共3个输入文件。
备注
我们必须在 unit_cell.str 中提供真实的原胞信息(即在满足平移周期性条件下具有最小体积的胞,否则无法正确分析不等价相互作用)。
cell.str 文件需要提供适当大小的超胞信息——这个超胞将用于四态法的第一性原理计算,因此不能太大(否则第一性原理计算的计算量太大);另外由于第一性原理计算时通常采用以超胞为边界的周期性边界条件,因此这个超胞必须足够大,才能够有效分辨我们所考虑的截断距离内的各个不等价相互作用的贡献。本节后面将会提到超胞是否足够大的测试方法。获取所需超胞的 cell.str 文件的方法参见3.1.3.8节(该节也是以 \(BiFeO_{3}\) 为例的)。
本模块因计算耗时短,暂不支持并行运算。
PASP.input 文件中一般需要根据不同体系修改的设置项包括“
block Z_values”、“block isaAB”、“block Spin_values”、“block magnetic_or_ligand”和“block fig.midst”,相应的部分内容如下所示。
#=========================================#
Model find_J_path_sym
# 元素原子序数,对应于cell.str
%block Z_values
83 26 8
%endblock Z_values
# 相互作用的元素编号(顺序见block Z_values)
%block isaAB
2 2
%endblock isaAB
# 自旋量子数的二倍
%block Spin_values
0 3 0
%endblock Spin_values
# 1磁性原子, 2配位原子,其他为0。Fe是磁性原子,O是配位原子。
%block magnetic_or_ligand
0 1 2
%endblock magnetic_or_ligand
# 截断距离(单位为Bohr)
%block fig.mdist
1 15
%endblock fig.mdist
#=========================================#
使用上述三个输入文件,运行PASP,在标准输出中(可在运行命令后面添加“ >log.out ”使其输出到 log.out 文件中)可以找到如下内容(#号后面为解释说明):
1 Atoms: 17 28 SE_SSE: 0 # 第1近邻的代表对是17和28号原子
pair_dist: 3.980978665681 # 第1近邻的距离(单位埃)
pair_coord: 0.101340529196 0.054885726174 0.037939863776 -0.165229314442 -3.212511228643 -2.220654565226 # 代表对原子的笛卡尔坐标
类似地,在设定的截断距离下共找到5种不等价相互作用,第二至五近邻的距离依次是5.492259242233、5.764360513360、6.783295859701和6.783295859701。其中第四和第五近邻距离相等,这有两种可能的原因:第一种,是因为它们虽然距离相等但是环境不等价,这时分析的结果是正确的;第二种,是因为超胞取的不够大导致的错误。为了判断是哪种原因,需要再生成充分大超胞(例如5×5×5)的 cell.str ,再用相同的 unit_cell.str 和 PASP.input 作为输入文件,重新运行PASP软件进行测试(参见文件夹 “example_public/find_J_path_sym/BiFeO3_555cell” 内容)。用5×5×5超胞测试时,可发现第五近邻距离增大到7.220732855773,这说明之前2×2×2超胞的结果是错误的,其原因是选取的超胞太小;反之,假如超胞足够大,而第四、第五近邻的距离值仍然相等,则结果是可靠的。经测试,3×3×3超胞即可得到正确的分析结果,因此在使用四态法计算前五近邻的 \(J\) 值时,至少需要使用3×3×3超胞;为了稳妥起见,一般需要取的更大一些,例如取4×4×4或4×4×5超胞。
2.1.2.3 参数计算与后续处理¶
根据2.1.2.2节内容,选定合适大小的超胞后,可以获得不同近邻代表对的原子编号,这里的编号即为 cell.str 中的原子编号。注意这里原子编号顺序与利用 post_VASP 脚本扩胞时生成的 POSCAR.multi 一致(参见3.1.3.8节内容),而 POSCAR.multi 可直接重命名为POSCAR作为VASP(常用的第一性原理计算软件)的输入文件之一。根据具体情况,自己写个脚本,在INCAR(VASP输入文件之一)的 MAGMOM 和 M_CONSTR 设置项中适当修改对应两个编号的原子的自旋取向,即可使用四态法计算得到所需的各个 \(J_{m}\) (或者 \(\mathbb{A}_{n}\) 和 \(\mathbb{J}_{m}\) )值。
为了进行后续的蒙特卡洛模拟(MC),我们还需要根据这些参数信息以及各代表对的坐标信息,填写 spin_exchange.dat 文件(格式要求参见3.3节),这是在MC步骤中需要用到的输入文件。在本例中,如果保留前五近邻的 \(\mathbb{J}_{m}\) 、前两近邻的 \(B_{m}\) 以及唯一的一种 \(\mathbb{A}_{n}\) ,可以按如下格式填写 spin_exchange.dat 文件内容(#号开头的首尾两行不算在文件内容之内):
#=========================================#
6 #共几组相互作用 #以下三行提供unit_cell.str的晶格信息
5.76436036846 -0.001063269563 -0.00073498713
3.14874999168 4.82837706456 -0.00073498713
3.14874999168 1.7053535363 4.5171888309
1 矩阵(9个数) 2 2 0.10134 0.05489 0.03794 0.10134 0.05489 0.03794
2 矩阵(9个数) 2 2 0.10134 0.05489 0.03794 -0.16523 -3.21251 -2.22065
3 矩阵(9个数) 2 2 0.10134 0.05489 0.03794 -2.51427 4.88433 0.03794
4 矩阵(9个数) 2 2 0.10134 0.05489 0.03794 5.86570 0.05382 0.03720
5 矩阵(9个数) 2 2 0.10134 0.05489 0.03794 -5.39645 3.32335 2.29727
6 矩阵(9个数) 2 2 0.10134 0.05489 0.03794 6.13227 3.32122 2.29580
0 # no dJ/du, i.e., no spin-lattice interaction
0 # single site anisotropy
0 # DM
2 #共几组四阶点乘形式相互作用
1 1 值(1个数) 2 2
0.10134 0.05489 0.03794 -0.16523 -3.21251 -2.22065
2 1 值(1个数) 2 2
0.10134 0.05489 0.03794 -2.51427 4.88433 0.03794
#=========================================#
请务必参照3.3节内容理解本文件各项数据的含义。注意这里并没有给出各个参数的具体数据,需要根据实际计算结果逐项填写(单位eV)。
2.1.3 蒙特卡洛模拟与CG局域优化 (Model Spin_lattice_interaction)¶
“Model Spin_lattice_interaction”是只适用于磁性体系的蒙特卡洛模拟模块,支持考虑公式 (4) 或公式 (5) 形式的相互作用;对于更复杂的相互作用形式,则需使用2.2.3节的“Model atomic_effective_H”。本节的“Model Spin_lattice_interaction”虽然通用性稍差,但因计算效率显著高于“Model atomic_effective_H”,因此经常用于处理不太复杂的磁性体系。
PASP中使用MC时,通常采用并行退火蒙特卡洛(parallel tempering Monte Carlo, PTMC)方法 11 12 13 。其中使用M个具有不同温度(即与不同热源接触)、彼此无相互作用的并行副本,并假定每个副本具有相同形式的哈密顿量 \(H(X_{m})\) ,这里用 \(X_{m}\) 表示第m个副本的全部自变量. 为讨论方便起见,不妨设各副本温度降序排列,换言之, \(\beta_{m}=1/(K_{b} T_{m})\) 按升序排列。
在PTMC模拟过程中,交替完成以下两种操作:一、每个副本同时而独立地各自按Metropolis算法进行正则系综的马尔科夫过程模拟;二、相邻温度 \(\beta_{m}\) 与 \(\beta_{m+1}\) 的两副本按概率 \(W\left(X_{m},\beta_{m} \lvert X_{m+1},\beta_{m+1}\right)\) 交换构型,要使得各副本都趋于平衡分布,可令
其中
采用这种PTMC方法可更快地离开局域极小值,并且显著减少模拟达到热平衡所需的弛豫时间。
在PTMC方法中,各副本的温度设置至关重要。使用更多低温的副本有利于找到基态但会增加达到热平衡所需时间;而研究比热容、磁化率等随温度的变化情况从而研究相变性质时,则需要较多的高温副本。常用的两种副本温度取法是: \(\left\{T_{m}\right\}\) 取为等差数列,或者 \(\left\{\beta_{m}\right\}\) 取为等差数列(分别对应 MC.input 中“ PT.Tmesh ”取2和1)。
本节换用 \(MnGe\) 作为例子,示例输入文件见文件夹 “example_public/Spin_lattice_interaction/MnGe/input” 。其中包括5个输入文件: PASP.input 、 MC.input 、 cell.str 、 unit_cell.str 和 spin_exchange.dat 。
如果读者希望沿用 \(BiFeO_{3}\) 的例子,只需参照2.1.2.3节填写 spin_exchange.dat 文件,再仿照 \(MnGe\) 的例子填写其他输入文件即可,注意MC阶段的扩胞数通常大于计算相互作用参数时的扩胞数。
备注
当模型中假定自旋归一化或采用实际自旋大小时,除了 spin_exchange.dat 文件的数据会有成比例的变化外, PASP.input 文件中的“
block Spin_values”以及 MC.input 中“block ExternalMagneticField”的设置方式也会有所不同。详见3.1.2.5节讨论。本模块支持并行,但须注意 MC.input 中PT.M必须是进程数的整数倍,否则软件会报错停止!
应根据蒙特卡洛模拟的实际需要,利用“
block SuperCell”扩胞(基于 cell.str 的结构扩胞);而 cell.str 既可以取原胞,也可以取适当扩胞的结构;当需要较准确地计算热容等统计量时,需使用充分大的超胞(这里的充分大,可以通过逐渐增加扩胞数,查看统计量的收敛趋势判断)。扩胞数建议先取较小值测试,根据计算耗时和模拟所得物理量的收敛情况再酌情增加扩胞数。此外,初步测试时, MC.input 中
PT.nblk、nbeg_blk、nblk不宜过大,否则测试时间可能过长。在初步测试顺利通过后,再酌情增加计算量。在增加到足够大的计算量时,这三个设置的推荐值是4000、2000、500。MC.input 中的“
spin_lat.relax_str”设置项决定在MC之后是否执行CG(共轭梯度下降法)局域优化。如果有特殊需要,也可以跳过MC步骤(“PT.nblk_T”设为0),直接执行CG局域优化。CG优化的意义在于,MC过程中产生各个状态具有随机性,不能保证MC过程中的最低能量态就是真正的最低能量态,甚至不能保证是局域最优的;而CG步骤能够将MC过程中的最低能量态优化变成局域最优态,不过需要注意这仍然不能保证是全局最优的。默认情况下,初态是随机产生的。如需指定初态,请参见2.1.3.1节内容。
本例中 spin_exchange.dat 文件采用的是更具通用性的矩阵形式的相互作用参数,如果需要采用更简洁的标量形式,格式与相应的设置项调整参见2.1.3.2节内容。特别注意,当需要使用CG功能时,只允许使用矩阵形式(否则会报错终止),且SIA、DM等相互作用信息也只能以矩阵形式提供(否则会报错终止),只有四阶点乘相互作用部分是不采用矩阵形式的!
PASP.input 和 MC.input 中经常修改的几个重要设置项列举如下。
#=========================================#
Model Spin_lattice_interaction
#基于cell.str结构扩胞
%block SuperCell
4 0 0
0 4 0
0 0 4
%endblock SuperCell
#各元素的原子序数
%block Z_values
25 32
%endblock Z_values
#各元素原子磁矩值(单位muB);若模型中自旋归一化,磁性原子需设为2
%block Spin_values
2 0
%endblock Spin_values
#研究哪两个编号的元素之间的磁相互作用,必须提供
%block isaAB
1 1
%endblock isaAB
#截断距离(单位Bohr),必须提供
%block fig.mdist
1 20
%endblock fig.mdist
Read_init_spin F #是否读取初始磁矩信息。若T,读取spin3.in构型
spin_lat.relax_str T #是否CG优化(仅Model Spin_lattice_interaction支持)
PT.M 64 #不同温度个数。注意必须是并行计算进程数的整数倍!
PT.low_temp 0.000001 #最低温,Hartree单位,0.00001约等于3.15 K
PT.high_temp 0.002 #最高温
PT.Tmesh 2 #温度值分布:1反比(1/T均匀),2正比(T均匀)
PT.nblk_T 400 # number of exchange steps
nbeg_blk 200 # number of exchange steps before measurement
nblk 50 # number of MC block steps. Default 10. 建议取10的整数倍
# 外磁场,沿z方向添加1 T磁场时设置为0 0 1(若自旋归一化,有效值为1/S)
#%block ExternalMagneticField
#0 0 1
#%endblock ExternalMagneticField
spin_lat.general_J T ### matrix form of J and SIA
spin_lat.LJmatSym T ### matrix form of J and SIA
#=========================================#
输出文件 C_PTMC_K.dat 给出不同温度(即设定的若干副本的温度)下的热容统计值,可用于判断相变温度。其中第一列为温度(单位K),第二列为热容(某种约化后的单位,只有相对大小有意义)。如果热容呈先上升后下降的趋势,则拐点处温度为相变温度(误差导致的局域扰动不算)。
输出文件 lowest_onlyspin.xsf 提供MC所得最低能量态的自旋组态信息,该文件可直接拖到vesta软件查看(可自动显示各个磁性原子的自旋矢量)。示例文件如下:
#=========================================#
CRYSTAL
PRIMVEC #以下给出超胞基矢
20.209247418 0.000000000 0.000000000
0.000000000 18.188322676 0.000000000
0.000000000 0.000000000 18.188322676
PRIMCOORD
256 1 #超胞中共256个磁性原子
21 3.254138469 0.727989657 1.798169154 -0.541570455 -0.565704786 -0.621835619 #21;笛卡尔坐标;自旋矢量
21 0.727989657 1.798169154 3.254138469 -0.544990535 -0.564932114 -0.619545821
21 1.798169154 3.254138469 0.727989657 -0.544077591 -0.562272638 -0.622759228
21 4.324317965 4.324317965 4.324317965 -0.543560942 -0.564108032 -0.621549379
21 8.306447946 0.727989657 1.798169154 -0.541579971 -0.565689152 -0.621841554
……(省略若干行)
21 19.481246397 19.481246397 19.481246397 -0.543558958 -0.564116670 -0.621543274
#=========================================#
该文件依次给出超胞中所有磁性原子的坐标和自旋矢量信息。每行开头的“21”是元素符号(代表 \(Sc\) 元素),这里总是输出“21”,与实际磁性原子的元素无关。另外,该文件中略去了所有的非磁性原子信息,这是为了在vesta中查看时排除干扰信息。输出的自旋矢量的模长正比于“ block Spin_values ”中磁性原子设置值,该设置值为2时输出的自旋模长是归一化的。顺便一提,该输出文件的晶格常数以及相应的原子坐标可以整体乘一个系数,用关键词“ write_only_spin.scale ”设置,例如“write_only_spin.scale 0.9”,缺省值为1.0(需要与Xcrysden软件搭配使用时,可能会需要设置成0.9)。
输出文件 spin3_lowest.out 以另一种格式给出MC所得最低能量态的自旋组态信息,示例如下:
#=========================================#
1 -0.54157046 -0.56570479 -0.62183562 #原子编号;归一化的自旋矢量
2 -0.54499053 -0.56493211 -0.61954582
3 -0.54407759 -0.56227264 -0.62275923
4 -0.54356094 -0.56410803 -0.62154938
5 0.00000000 0.00000000 0.00000000
6 0.00000000 0.00000000 0.00000000
7 0.00000000 0.00000000 0.00000000
8 0.00000000 0.00000000 0.00000000
9 -0.54157997 -0.56568915 -0.62184155
……(省略若干行)
508 -0.54355896 -0.56411667 -0.62154327
509 0.00000000 0.00000000 0.00000000
510 0.00000000 0.00000000 0.00000000
511 0.00000000 0.00000000 0.00000000
512 0.00000000 0.00000000 0.00000000
#=========================================#
该文件每行第一个数字为超胞中原子编号,第二至四个数为相应的自旋矢量。可以看到该文件中各磁性原子的出现顺序与 lowest_onlyspin.xsf 一致,只是该文件多出了非磁性原子的磁矩信息。该文件中输出的磁性原子自旋矢量总是归一化的(而非磁性原子的自旋矢量总是0)。
如果采用了CG优化,则还会产生一组加了 relax-前缀 的文件,提供相应的CG优化后的自旋组态信息。例如对应前面提到的 lowest_onlyspin.xsf 和 spin3_lowest.out 文件,会产生CG优化后的 relax-lowest_onlyspin.xsf 和 relax-spin3_lowest.out 文件。
此外,如果不止关心最低能量态,还关心不同温度下的随机状态,可以查看 T_*_final. (*部分为温度值,单位Hartree)开头的若干xsf文件,它们提供了各个温度副本的MC末态信息。文件名后半部分有两种,第一种是 onlyspin.xsf ,仅提供磁性原子信息;第二种是 all_atoms_spin.xsf ,会提供所有原子的自旋矢量信息。
2.1.3.1 设置初始构型与计算指定构型的能量¶
如需自定义蒙特卡洛模拟的初始构型,需要在 MC.input 中将如下设置项修改为T:
Read_init_spin T #是否读取初始磁矩信息。若T,读取spin3.in构型
在本节所选Model中,通过 spin3.in 文件提供初始构型,即超胞中各原子的自旋矢量信息。该文件格式与输出文件 spin3_lowest.out 一致(参见前文示例),需要依次提供超胞中各个原子的编号和自旋矢量的三个分量。哪些编号的原子是磁性原子,哪些是非磁性原子,可以在 spin3_lowest.out 文件中查得;各磁性原子的坐标,可依次在 lowest_onlyspin.xsf 文件中查得(如果一开始缺少 spin3_lowest.out 和 lowest_onlyspin.xsf 文件,可以先不设置初始构型,用很少的步数完成一次蒙特卡洛模拟,以获得这两个输出文件)。
如果需要直接获取初始构型的能量(在 spin_exchange.dat 设定的有效哈密顿量模型下的预测能量),可以在 MC.input 中将“ PT.nblk_T ”设置为0(即不进行蒙特卡洛模拟步骤),并且将“ spin_lat.relax_str ”设置为F(即不进行CG优化),运行PASP时可以在标准输出(运行软件时可通过“ > log.out ”将标准输出的内容输出到 log.out 文件中)中找到如下相应内容:
Node: 0 Initial Energy (in Hartree) is: -3.985021379812
Node: 0 Initial Energy (in eV) is: -108.439015000000
其中依次给出了Hartree单位和eV单位的初态能量。
2.1.3.2 提供非矩阵形式参数的情形¶
前面的示例文件中假定两体相互作用参数都有矩阵形式提供。若体系不考虑SOC,则两体相互作用可以简单地用各向同性的标量J描述,相当于J矩阵采用了对角元相等的对角化形式。此时, PASP.input 的以下两行设置项需要注释掉(使之无效,而缺省值为F):
#spin_lat.general_J T ### matrix form of J and SIA
#spin_lat.LJmatSym T ### matrix form of J and SIA
直接设置成F也是等效的:
spin_lat.general_J F ### matrix form of J and SIA
spin_lat.LJmatSym F ### matrix form of J and SIA
此时,参见3.3节内容, spin_exchange.dat 文件第一部分数据的“每行内容”中“J矩阵(逐行填写矩阵元,共9个数)”简化为“J值(仅1个数)”,其他部分内容都不变。本节(2.1.3节)前面提到的 \(MnGe\) 的示例输入文件 spin_exchange.dat 在该情形下可以相应地改成:
#=========================================#
2 #共几组相互作用 #以下三行提供unit_cell.str的晶格信息
5.0523118545661250 0.0000000000000000 0.0000000000000000
0.0000000000000000 5.0523118545661250 0.0000000000000000
0.0000000000000000 0.0000000000000000 5.0523118545661250
1 -0.02897 1 1 1.79817 3.25414 0.72799 0.72799 1.79817 3.25414
2 0.00622 1 1 0.72799 -3.25414 -1.79817 4.32432 -0.72799 -0.72799
0 # no dJ/du, i.e., no spin-lattice interaction
0 # single site anisotropy
0 # DM
0 #共几组四阶点乘形式相互作用
#=========================================#
以上由矩阵形式化为标量形式时,是通过取对角元平均值得到的;也可直接通过无SOC的四态法直接计算得到。
2.2 一般性有效哈密顿量方法¶
2.2.1 一般性有效哈密顿量的形式和构建 (Model Invariant)¶
磁电体系的一般性有效哈密顿可以写为 \(H=H_{spin}+H_{lat}+H_{spin-lat}\) ,其中 \(H=H_{spin}\) 为有效自旋哈密顿, \(H_{lat}\) 为晶格部分的有效哈密顿,而 \(H_{spin-lat}\) 为自旋-晶格耦合相互作用。只用 \(H=H_{spin}\) 可以得到磁性体系的基态和有限温度性质,只用 \(H=H_{lat}\) 可以研究非磁性铁电体系的基态和有限温度性质,而包含自旋-晶格耦合相互作用的完整哈密顿才可以用于研究多铁性体系。类似于合金研究里的团簇展开方法,有效自旋哈密顿可以表示成不同自旋团簇贡献的和: \(H_{spin}=\textstyle\sum_{k}H_{k}\) , \(H_{k}\) 为 \(k\) -体相互作用贡献,如 \(H_{2}=\textstyle\sum_{<i,j>}J_{p_{i},q_{i},r_{i},p_{j},q_{j},r_{j}}^{ij}S_{ix}^{p_{i}}S_{iy}^{q_{i}}S_{iz}^{r_{i}}S_{jx}^{p_{j}}S_{jy}^{q_{j}}S_{jz}^{r_{j}}\) ,其中 \(p_{i},q_{i},r_{i},p_{j},q_{j},r_{j}\) 为非负整数,且 \(p_{i}+q_{i}+r_{i}\ge 1\) 和 \(p_{j}+q_{j}+r_{j}\ge 1\) ,同时为了使能量在时间反演下 \(\left(S_{i\alpha}\to- S_{i\alpha}\right)\) 不变, \(p_{i}+q_{i}+r_{i}+p_{j}+q_{j}+r_{j}\) 必须是偶数。可以看出 \(H_{2}\) 包含了通常的海森堡相互作用 \(\widetilde{J}^{ij}S_{i}\cdot S_{j}\) 。如果体系不存在自旋轨道耦合,或自旋轨道耦合可以忽略时,自旋相互作用项在所有可能的转动操作下不变,即只包含自旋-自旋点乘项,如 \(\boldsymbol{S}_{i} \cdot \boldsymbol{S}_{j}\) 和 \(\left(\boldsymbol{S}_{i} \cdot \boldsymbol{S}_{j}\right)\left(\boldsymbol{S}_{k} \cdot \boldsymbol{S}_{l}\right)\) 。另外,对于具体材料,由于空间群对称性,可能可以进一步简化,比如当自旋对的中心正好是空间反演中心时,该自旋对不存在DM相互作用。由于可能包含的项非常多,找出其中非零不等价的自旋相互作用项比较困难。PASP采用系统的群论方法解决这个问题:对于某个选定的自旋团簇,先确定该自旋团簇的点群对称性;因为这个自旋团簇的非零不等价的自旋相互作用项一定是该自旋团簇点群的恒等表示的基,PASP可以采用投影算符方法把这些非零不等价的自旋相互作用项找出来。
对于晶格部分的有效哈密顿,它一般可表示为局域声子模式 \(u\) 和应变 \(\eta\) (包括均匀应变和非均匀应变)的函数: \(H_{l a t}=H_{l a t}(u, \eta)\) ,其中可能包括局域声子模式的自能项(如 \(u_{i}^{2}\) 和 \(u_{i}^{4}\) ),短程的局域声子模式之间的相互作用(如 \(u_{i} u_{j}\) 和 \(u_{i}^{2} u_{j} u_{k}\) ),长程的电偶极-电偶极相互作用,只与应变相关纯弹性能,以及局域声子模式和应变的相互作用项(如 \(u_{i} \eta_{i}\) )。一般而言,短程的局域声子模式之间的相互作用、局域声子模式和应变的相互作用可能存在的项非常多,同样,PASP可以利用群论方法找出所有可能(给定阶数和相互作用距离内)不等价的相互作用项。
有效哈密顿的自旋-晶格耦合相互作用可表示为自旋 \(S\) ,局域声子模式 \(u\) 和应变的函数 \(\eta\) ,即 \(H_{\text {spin-lat }}=H_{\text {spin-lat }}(S, u, \eta)\) ,比如, \(H_{\text {spin-lat }}\) 可能包括 \(\left(\boldsymbol{S}_{i} \cdot \boldsymbol{S}_{j}\right) u_{m}\) 和 \(S_{i x} S_{j y} u_{m}^{2} \eta\) 。为了使能量在时间反演下不变,其中自旋项的总阶数必须是偶数。若不考虑自旋轨道耦合效应,或由于点群对称性的存在,相互作用项还可进一步简化。同样,PASP可利用群论方法找出 \(H_{\text {spin-lat }}\) 中所有可能(给定阶数和相互作用距离内)不等价的相互作用项。
除此之外,在合金材料中,还可以涉及元素种类的自由度;当晶体材料中含有有机分子时,还可能涉及有机分子的取向、位移等自由度;此外还可以考虑电荷量、轨道序等自由度。一般地,总的有效哈密顿量可以采用这些变量的泰勒展开形式,再根据时间反演不变以及点群对称性等条件淘汰或合并一些项,从而给出所有可能(给定阶数和相互作用距离内)不等价的相互作用项。
总之,当我们采用各自由度的泰勒展开形式并适当淘汰、合并项之后,模型总可以抽象成如下线性组合的形式:
其中 \(\left\{h_{j}\right\}\) 为线性组合的基函数(basis function),这部分是构型的函数,对应于各种不等价相互作用形式,一般为多项式形式; \(\left\{C_{j}\right\}\) 为线性组合的待定系数,这是与构型无关的相互作用参数。有效哈密顿量中不重要的常数项对应于 \(h_{1} \equiv 1\) 。
在PASP的“Model Invariant”模块中,可以自定义需要研究哪些自由度,然后通过相应的对称性分析,判断在指定的截断阶数和截断距离下有哪些不等价的相互作用,即给出各个 \(\left\{h_{j}\right\}\) 的具体表达式。当指定具体构型(包含自旋组态、局域声子模式和应变等信息)时,可以计算得到 \(\left\{h_{j}\right\}\) 的取值,再利用第一性原理计算可以计算体系总能量,作为 \(H_{eff}\) 的取值。当我们计算多组构型的数据时,用上标 \(^{(i)}\) 表示构型的编号,则可以利用PASP以及第一性原理计算,得到多组 \(\left\{\left\{h_{j}^{(i)}\right\}, H_{e f f}^{(i)}\right\}\) 数据。我们后续将利用这些数据,筛选出重要相互作用项(将不重要的若干 \(\left\{C_{j}\right\}\) 取为0),再拟合得到各个重要的 \(\left\{C_{j}\right\}\) 值(详见2.2.2节),从而完成构建有效哈密顿量的步骤。之后即可利用这个有效哈密顿量进行蒙特卡洛模拟(2.2.3节)、CG局域优化(2.2.3节)或者分子动力学模拟(2.2.4节)。
备注
这里计算的 \(\left\{h_{j}^{(i)}\right\}\) 是平均到“每个原胞”的效果,而VASP等第一性原理计算给出的总能量通常是平均到“每个超胞”的效果。为了解出正确的 \(\left\{C_{j}\right\}\) 值,需要将第一性原理计算结果除以“超胞中所含原胞数”的结果视为 \(H_{e f f}^{(i)}\) 。在解出 \(\left\{C_{j}\right\}\) 值后,对于任意给定的新构型,可以用PASP分析得到它的 \(\left\{h_{j}\right\}\) ,根据公式 (14) 将其与 \(\left\{C_{j}\right\}\) 点乘,得到平均到“每个原胞”的总能量预测值。
在该Model中,软件总是假定自旋矢量模长为1(无量纲),另外局域声子模式和应变的大小也作适当的无量纲化处理,从而使得所有相互作用系数单位均与能量的单位一致(默认eV单位)。
自定义研究哪些自由度,主要通过 PASP.input 的“block local_mode.mode_definition”完成(详见3.1.7.5节),目前在本手册的示例中仅涉及自旋和局域声子模式两种自由度;另外应变自由度直接通过“
Invariant.strain_power”和“Invariant.pure_strain_power”设定考虑到几阶,从而决定是否考虑该自由度(设为0阶即为不考虑该自由度)。以下将针对无SOC时的磁性体系(相互作用仅需考虑点乘形式)、考虑SOC时的磁性体系(相互作用采用一般形式)、铁电体系三种常见情形,依次给出三组示例(分别在2.2.1.1节、2.2.1.2节和2.2.1.3节)。当需要综合考虑自旋、局域声子模式时,只需要在“block local_mode.mode_definition”中混合定义两类自由度即可。通常超胞不能太小,建议保证超胞的最短基矢不小于两倍截断距离,否则在周期性边界条件影响下,部分相互作用的贡献将混在一起难以分辨。为了避免大超胞导致过大的计算量,我们也可混合使用多种不同大小的超胞数据,以便在总计算量不太大的前提下仍然能够有效分辨各个相互作用的贡献。当混合使用多种不同大小的超胞时,需要注意,对于每个构型,除了要用第一性原理方法计算它的能量,还要在相同条件下计算它所采用的超胞的参考态的能量(通常取最简单、对称性最高的态作为参考态,如铁磁态、顺电态),用构型能量减去参考态能量,再除以超胞所含的原胞数,作为它的 \(H_{e f f}^{(i)}\) 数据。
本模块暂不支持并行。
为了产生多组 \(\left\{\left\{h_{j}^{(i)}\right\}, H_{e f f}^{(i)}\right\}\) 数据,主要有两类思路。第一类思路是自己编写脚本,按自己指定的规则(通常是完全随机,或者在一些限制条件下随机)生成若干构型,再分别用PASP分析它们的 \(\left\{ h_{j}^{(i)}\right\}\) ,以及通过第一性原理计算得到它们的 \(\left\{ H_{e f f}^{(i)}\right\}\) ,其中运行PASP时每次仅分析其中一种指定构型的 \(\left\{h_{j}^{(i)}\right\}\) ,需要通过多次循环调用PASP来分析所有构型的 \(\left\{h_{j}^{(i)}\right\}\) ;第二类思路是直接利用PASP随机生成若干构型(通常局域声子模式和应变自由度是在指定取值范围内完全随机,而自旋自由度总是完全随机取向),这时只需调用一次PASP就能产生所有的构型信息和 \(\left\{h_{j}^{(i)}\right\}\) ,之后再利用这些构型信息完成多组第一性原理计算。第一类思路对应 PASP.input 中“ local_mode.cal_para_method ”设置值取6(例如2.2.1.1节和2.2.1.2节示例)或5,第二类思路则对应设置值取1(例如2.2.1.3节示例)。三种设置值的含义和差别详见3.1.7.7节。实际使用时需要根据具体需求来决定采用哪种设置值。
2.2.1.1 磁性-点乘形式¶
这里先以“ local_mode.cal_para_method ”设置值为6的情形为例介绍使用方法及输出文件;对于其他设置值的情况,将在本节最后简单介绍。
本节以 \(TbMnO_{3}\) 为例,保留到四阶的点乘相互作用项。为了计算某个给定的自旋组态对各种相互作用的贡献,所需的全部输入文件在 example_public/Magnetic_noSOC_TbMnO3/input 文件夹下,包括 unit_cell.str 、 cell.str 、 PASP.input 、 distortion.pos 和 distortion.MAGMOM 共5个输入文件。
备注
若在 cell.str 中提供的是原胞的信息,可略去 unit_cell.str 文件。例如在本组示例文件中 unit_cell.str 原则上可以省略。
distortion.pos 文件与POSCAR格式一致,提供超胞信息,本例中是2×2×2的超胞。第一行作为备注信息不重要;第六行可以不写化学符号,直接写各元素原子个数,也可以先写一行化学符号再在下一行写各元素原子个数。参见3.1.3.8节内容,可以使用 post_VASP 脚本,基于POSCAR文件扩胞获得 POSCAR.multi 文件,再将其重命名为 distortion.pos 即可。
distortion.MAGMOM 文件,需按照 distortion.pos 中的原子顺序,依次给出超胞中各个原子的磁矩的x、y、z分量。磁矩大小不重要,软件内部会归一化处理。
PASP.input 中的几个经常修改的重要设置项列举如下。
#=========================================#
Model Invariant
#各元素的原子序数(无需额外补充Fe元素)
%block Z_values
65 25 8
%endblock Z_values
%block Spin_values #磁性原子写2(蒙卡时必须取2,这里暂无影响),其他写0
0 2 0
%endblock Spin_values
Invariant.power 4 #default 4, the max power of invariant,最大阶数
Invariant.strain_power 0 #default 1, the power of strain, 应变部分最大阶数
Invariant.pure_strain_power 0 #default 2, the power of pure strain,纯应变项最大阶数
fig.maxk 4 # the max power of many body interation.最多几体
%block fig.mdist
1 15 15 15 ### Bohr单位,不同体数的截断距离
%endblock fig.mdist
local_mode.cal_para_method 6 # 6 是从distortion.pos/MAGMOM中读取构型
local_mode.read_interaction F #是否从local_mode_PM.dat中读取相互作用信息
%block local_mode.mode_definition
2 # method, 2 is the general one, as below
12 # num of modes
1 spin
2
2.646572590 0.000000000 0.000000000 1.0 0.0 0.0
…………(本设置项行数过多,此处略去部分内容)
%endblock local_mode.mode_definition
Invariant.SOC F ### default T。是否考虑SOC
local_mode.GSM2lm_int T #采用点乘形式时需要此行
GSM.maxpower 2 #采用点乘形式时需要此行,最大阶数为该设置值*2
#=========================================#
使用上述5个输入文件,运行PASP可得到输出文件 distortion.ene_symbol ,以及 spin_cluster.dat 、 all_interaction_type_Spin_xyzform.dat 和 local_mode_PM.dat 等介绍各相互作用项的详细信息的文件。
其中输出文件 distortion.ene_symbol 给出了不等价相互作用个数(第一行“#npara”前面的数字)以及给定的自旋组态对各种相互作用的总贡献(平均到每个原胞,单位eV)。例如内容如下:
#=========================================#
346 #npara #总的不等价相互作用个数
1 0.243841635894 #第1种;自旋组态对该作用的贡献值(每原胞)
2 1.379560781428 #第2种;自旋组态对该作用的贡献值(每原胞)
3 0.550369143339
……(省略若干行)
346 -1.060482829187
#=========================================#
每种不等价相互作用的含义可在 all_interaction_type_Spin_xyzform.dat 、 local_mode_PM.dat 和 spin_cluster.dat 中读到。其中 spin_cluster.dat 可读性最强,但仅适用于本节情形(采用点乘形式相互作用的磁性体系);另外两个文件则适用于一般情形。
先介绍 spin_cluster.dat 输出文件,该文件中可查得每种不等价相互作用的表达式(以点乘形式描述)。例如第12种不等价相互作用的相关信息为:
#=========================================#
fractional coordination of the 6 cluster: #第6种代表簇(或代表对)
0.500000017003 0.000000000000 0.000000000000
0.500000017003 1.000000000000 0.000000000000
npair: 1
1 : 1 2
nindep: 2
11 -th independent parameter 2 6
1 : 1
Readable term: (S1.S2)^1
12 -th independent parameter 2 6 #第12种相互作用;2体第6种团簇
2 : 2
Readable term: (S1.S2)^2 #第12种相互作用形式
#=========================================#
即第12种不等价相互作用是二体四阶相互作用项,代表的原子对的坐标在“fractional coordination of the 6 cluster”后面两行给出,注意这里的坐标是以“原胞基矢”作为单位的,即采用了分数坐标!
另外, all_interaction_type_Spin_xyzform.dat 输出文件的部分内容示例如下:
#=========================================#
icluster, #sites, which-cluster: 3 2 1
1 2 u_{1,Sx} u_{2,Sx} +u_{1,Sy} u_{2,Sy} +u_{1,Sz} u_{2,Sz}
2 4 u_{1,Sx}^2 u_{2,Sx}^2 + 2.0000u_{1,Sx} u_{1,Sy} u_{2,Sx} u_{2,Sy} + 2.0000u_{1,Sx} u_{1,Sz} u_{2,Sx} u_{2,Sz} +u_{1,Sy}^2 u_{2,Sy}^2 + 2.0000u_{1,Sy} u_{1,Sz} u_{2,Sy} u_{2,Sz} +u_{1,Sz}^2 u_{2,Sz}^2
#=========================================#
其中第一行三个数字分别表示:第3种原子簇(cluster),该原子簇包含2个原子(即二体相互作用),在二体相互作用中编号为1。第二行两个数字分别表示:第1种相互作用(与distortion.ene_symbol中的第几种一致),阶数为2。第二行后面给出了相互作用的数学形式(展开式,未作合并同类项等处理),该例中实际为 \(S_{1}\cdot S_{2}\) ,即海森堡相互作用。类似地,第三行是第2种相互作用,4阶,为双二次(biquadratic)相互作用 \(\left(S_{1}\cdot S_{2}\right)^{2}\) 。不过,在该文件中无法查看代表簇(这里为代表对)的坐标。
在 local_mode_PM.dat 输出文件中,第3种原子簇对应的部分相关内容如下:
1#=========================================#
2======================================== #分段标志,每段为一种团簇
3 3 2 1 2 12 6 0 #Interaction,ik,inf,nint_para,nmode_all,nmode_cluster,nint_para_lamp
4 #第3种团簇:2体
5 #二体的第1种
6 #该团簇涉及的参数个数为2
7 #相关的总模式数
8 #不考虑应变的相关的模式数
9 #模式过大时的相关参数个数(这里总为0,不重要)
10 6 #max number of local modes per site #每个原子最多可能的相关模式数(不重要)
11#crd #代表簇(或代表对)的各原子坐标。注意是原胞基矢下的分数坐标!
12 1 0.000000000000 0.499999991436 -0.499999988788
13 2 0.000000000000 0.499999991436 0.000000000000
14#mode_ind依次列出相关的每种模式信息,这部分行数等于“相关的总模式数”。
15 1 7 1 #代表簇的第1个原子,编号7的模式,该原子的第1个模式
16 1 8 2
17 1 9 3
18 2 10 1
19 2 11 2
20 2 12 3
21 0 1 1 #代表簇的第0个原子是指与应变相关的模式
22 0 2 2
23 0 3 3
24 0 4 4
25 0 5 5
26 0 6 6
27#nmode_site 应变相关的模式数;各原子的相关模式数(几体就有几个数)
28 6 3 3
29#mode_site
30 0 #atom #代表簇的第几个原子。0代表应变相关
31 1 2 3 4 5 6 #应变依次对应哪些编号的模式
32 1 #atom #代表簇的第1个原子
33 7 8 9 #第1个原子依次对应哪些编号的模式
34 2 #atom #代表簇的第2个原子
35 10 11 12
36
37#int_para
38 1 1 2 3 12 0 F #ipara,ipara_gb,power,n,nmode,im_read,lonebody
39 #该代表簇的第1种参数
40 #总的参数编号1
41 #对应2阶相互作用
42 #表达式共3项
43 #与该参数可能相关的模式数为12(总是加上六种应变模式)
44 #后面的“0 F”不重要
45#terms #以下依次给出各项的信息(共3项)。
46 1 #j #第1项信息。以下各位数字依次是1、2号原子的xyz,6种应变模式
47 1 0 0 1 0 0 0 0 0 0 0 0 #表示S_1x*S_2x
48 2 #j #第2项信息
49 0 1 0 0 1 0 0 0 0 0 0 0 #表示S_1y*S_2y
50 3 #j #第3项信息
51 0 0 1 0 0 1 0 0 0 0 0 0 #表示S_1z*S_2z
52#coeff #依次给出各项的系数
53 1 1.000000000000 #j,value #第1项系数为1.0
54 2 1.000000000000 #j,value #第2项系数为1.0
55 3 1.000000000000 #j,value #第3项系数为1.0
56
57 2 2 4 6 12 0 F #ipara,ipara_gb,power,n,nmode,im_read,lonebody
58 #该代表簇的第2种参数
59 #总的参数编号2
60 #对应4阶相互作用
61 #表达式共6项
62 #与该参数可能相关的模式数为12
63#terms #共6项
64 1 #j
65 2 0 0 2 0 0 0 0 0 0 0 0 #表示S_1x^2*S_2x^2
66 2 #j
67 1 1 0 1 1 0 0 0 0 0 0 0 #S_1x*S_1y*S_2x*S_2y
68 3 #j
69 1 0 1 1 0 1 0 0 0 0 0 0
70 4 #j
71 0 2 0 0 2 0 0 0 0 0 0 0
72 5 #j
73 0 1 1 0 1 1 0 0 0 0 0 0
74 6 #j
75 0 0 2 0 0 2 0 0 0 0 0 0
76#coeff
77 1 1.000000000000 #j,value
78 2 2.000000000000 #j,value
79 3 2.000000000000 #j,value
80 4 1.000000000000 #j,value
81 5 2.000000000000 #j,value
82 6 1.000000000000 #j,value
83
84--Block for symmetrically equivalent clusters-- #后面列举对称等价的团簇(原子对)
85 4 #neq #等价的团簇数
86#后面的内容提供等价团簇的坐标,以及实际的相互作用形式,这里省略。
87#=========================================#
可以看出, local_mode_PM.dat 文件中给出的表达式与 all_interaction_type_Spin_xyzform.dat 内容一致。但 local_mode_PM.dat 文件进一步给出了代表簇的各原子坐标,同时给出了等价的团簇信息。
备注
关于“ local_mode.cal_para_method ”的不同设置情形,建议仔细阅读3.1.7.7节的相应内容。本段补充介绍在该设置项的几种选项下,输出文件的差别。在本节示例中采用了“ local_mode.cal_para_method ”为6的设置,其优点是可以根据实际需要自定义构型,更具灵活性。在设置为1时,自动生成的随机构型由 *.vasp_MAGMOM_spin 提供(* 表示编号,下同),其格式与 distortion.MAGMOM 一致,这两个文件中模长由“%block Spin_values”的设置值决定;另外 *.vasp_MAGMOM 与 VASP的INCAR 相应设置项( MAGMOM 和 M_CONSTR )格式一致;第一性原理计算时所需的POSCAR由 * .vasp提供;与 distortion.ene_symbol 对应的输出文件名变为 *.ene_symbol 。在设置为5时,生成的文件名与设置为1时类似,只是 *仅取1,因为此时仅生成一种自定义的构型。
2.2.1.2 磁性-一般形式¶
如果考虑SOC,则有效自旋哈密顿量的形式将不再局限于点乘的组合。本节沿用2.2.1.1节的例子(建议先仔细阅读2.2.1.1节再读本节),即以 \(TbMnO_{3}\) 为例,保留到四阶相互作用项,但因考虑SOC而不再局限于点乘形式相互作用。为了计算某个给定的自旋组态对各种相互作用的贡献,所需的全部输入文件在 example_public/Magnetic_SOC_TbMnO3/input 文件夹下。
此时大部分输入文件无需改变,只有 PASP.input 中需要设置“ Invariant.SOC T ”(缺省值即为T),同时去掉点乘形式时用到的设置“ local_mode.GSM2lm_int T ”和“ GSM.maxpower 2 ”(用#注释使之无效即可)。修改后的相应设置项如下所示:
#=========================================#
Invariant.SOC T ### default T。是否考虑SOC
#local_mode.GSM2lm_int T #采用点乘形式时需要此行
#GSM.maxpower 2 #采用点乘形式时需要此行,最大阶数为该设置值*2
#=========================================#
此时主要输出文件也与2.2.1.1节相同,只是不再产生 spin_cluster.dat 文件(因为该文件仅提供点乘形式的自旋相互作用信息)。经测试运行可发现共有11086种不等价相互作用,显著多于仅考虑点乘形式时的346种。
备注
如果需要更精确地指定哪几种相互作用考虑SOC,而其他类型的相互作用不考虑SOC(仅保留点乘形式),可以在“ block local_mode.special_SOC ”中具体设置,详见3.1.7.4节。
2.2.1.3 铁电¶
建议在读本节前,先熟悉2.2.1.1节内容。铁电体系与磁性体系在输入文件的主要差别在于“block local_mode.mode_definition”的内容,以及铁电体系常常需要考虑分析应变相关项。
关于“ local_mode.cal_para_method ”设置项,如果取6(通过 distortion.pos 文件自定义构型),由于铁电体系不用考虑自旋模式,此时无需再提供 distortion.MAGMOM 文件。而 distortion.pos 文件所提供的结构应当是考虑到局域声子模式所导致的原子位移之后的结构;注意软件从自定义构型(由distortion.pos定义)解出各局域声子模式大小时可能会出现解不唯一的情况,这时软件会自动找出各模式大小的平方和最小的解。分析所得的各局域模式大小信息会列在输出文件 distortion.str_mode_amp 中,该构型对各种相互作用的贡献会输出到 distortion.ene_symbol 文件(平均到每个原胞)。如果采用“ local_mode.cal_para_method ”为1的设置,则可以根据“ block local_mode.mode_range ”设定的各模式取值范围随机生成指定数量的结构(数量由“local_mode.nstr_random”决定),各结构信息输出到*.vasp( * 为编号,下同)文件(可作为VASP的POSCAR使用),各局域模式大小信息输出到 *.str_mode_amp 文件,各构型对各种相互作用的贡献输出到 *.ene_symbol文件 (平均到每个原胞)。
本节以 \(BaTiO_{3}\) 体系“ local_mode.cal_para_method ”取1的情形为例,用PASP直接生成100个随机构型(取2×2×2超胞)。所需的全部输入文件在 example_public/ Ferroelectric_user_defined_mode_BaTiO3/input 文件夹下。其中 cell.str 中提供的是原胞的信息,因此原则上可略去 unit_cell.str 文件。另外 seed.in 文件是为了固定随机数种子,以便重复结果,通常也可省略。
备注
相比于磁性体系,铁电体系常常需要考虑应变项,可通过“
Invariant.strain_power”和“Invariant.pure_strain_power”设置含应变项与纯应变项分别最大考虑到几阶,两项都取0时可以忽略应变相关项。虽然理论上讲,把每个原子的xyz三个位移自由度都考虑到才是最严格、最全面的,但那样会导致有效哈密顿量过于复杂,也大大增加了拟合参数的难度。所以通常我们仅考虑几种典型的局域集体位移模式,但需注意其中各原子的位移比例应该是恰当的,以保证我们使用定义的少量自由度能够有效描述该体系的几种典型状态(如顺电和铁电态)。在本例中局域模式是按如下步骤确定的:使用顺电态的原胞,仅允许各原子在z方向弛豫,用VASP软件找到能量最低的铁电态结构(沿001方向,而非111方向的铁电基态),我们这里是按这个001方向的铁电态结构定义的局域模式;先算出该铁电态结构中各原子相对顺电态的位移;每个位移值减去原胞中所有原子位移量的平均值(去掉平移对称性的影响);每个位移值再除以“该原子-局域模式中心原子(这里以Ti为中心原子)”的配位数(该例中O-Ti配位数为2,Ba-Ti配位数为8,所以O的位移值除以2,Ba的位移值除以8,而Ti的位移值不变);最后再对局域模式定义中涉及的所有原子的位移值的“总矢量”归一化(即总的位移平方和为1,单位Bohr2),至此可以确定局域模式涉及的各个原子的位移量(在 PASP.input 的“block local_mode.mode_definition”部分需要用到这些数据)。其中最后的归一化是可以省略的,因为软件内部会自动归一化。这样处理的效果是,无论怎样扩胞,当所有的沿z方向的局域模式都取适当的相同值时,可以构造出VASP软件所找到的001方向的铁电态结构。除了根据VASP弛豫结果构造局域模式外,也可以通过求解声子模式等方法构造。
需要注意“block local_mode.mode_range”所需的行数应当是“局域模式数+6”,这里是3+6=9。注意到在本例中“block local_mode.mode_definition”仅定义了一种局域声子模式,但是实际上分析出了3种局域声子模式(PASP软件根据对称性自动添加等价的模式)。由于可能难以事先预测PASP软件分析后总共有多少种局域模式,“block local_mode.mode_range”的正确行数可能难以一次性写对。为此,可以先测试运行一次,在测试运行中,将“block local_mode.mode_range”的内容注释掉(使用缺省设置,可以避免因为行数错误而报错),并将“
local_mode.nstr_random”暂设为1,展开阶数、截断距离等可以暂取较小值,看产生多少 local_mode-*.xsf 文件(这些文件给出了各个局域模式涉及的原子的原子序数、笛卡尔坐标以及各原子位移),这就是实际的局域模式数,这个数加上6就是“block local_mode.mode_range”所需的行数。PASP.input 中经常修改的重要设置项列举如下。
#=========================================#
Model Invariant
# local_mode.cal_para_method取1时,一般利用block SuperCell扩胞
%block SuperCell
2 0 0
0 2 0
0 0 2
%endblock SuperCell
#各元素的原子序数
%block Z_values
8 22 56
%endblock Z_values
Invariant.power 4 #default 4, the max power of invariant,最大阶数
Invariant.strain_power 1 #default 1, the power of strain, 应变部分最大阶数
Invariant.pure_strain_power 2 #default 2, the power of pure strain,纯应变项最大阶数
fig.maxk 4 # the max power of many body interation.最多几体
%block fig.mdist
1 15.25 15.25 15.25 ### Bohr单位,不同体数的截断距离
%endblock fig.mdist
%block local_mode.mode_range #各模式的最大取值(常用);分段数(不常用)
0.27592808 10
0.27592808 10
0.27592808 10
0.025 2
0.025 2
0.025 2
0.025 2
0.025 2
0.025 2
%endblock local_mode.mode_range
local_mode.cal_para_method 1 # 1随机生成; 6从distortion.pos等文件读取构型;
local_mode.nstr_random 100 #随机结构数(local_mode.cal_para_method取1时用)
local_mode.read_interaction F #是否从local_mode_PM.dat中读取相互作用信息
%block local_mode.mode_definition
2 # method, 2 is the general one, as below
1 # num of modes
15 lamp 0.0 2.76
2 1 1 1 1 1 1 3 3 3 3 3 3 3 3
2.01665571 2.01665571 2.01665571 0.0 0.0 0.84914560 #Ti
2.01665571 0.00000000 2.01665571 0.0 0.0 -0.12643559 #O1
4.03331143 2.01665571 2.01665571 0.0 0.0 -0.12643559 #O1
2.01665571 4.03331143 2.01665571 0.0 0.0 -0.12643559 #O1
0.00000000 2.01665571 2.01665571 0.0 0.0 -0.12643559 #O1
2.01665571 2.01665571 0.00000000 0.0 0.0 -0.31944756 #O2
2.01665571 2.01665571 4.03331143 0.0 0.0 -0.31944756 #O2
0.00000000 0.00000000 0.00000000 0.0 0.0 0.03693648 #Ba
4.03331143 0.00000000 0.00000000 0.0 0.0 0.03693648 #Ba
4.03331143 4.03331143 0.00000000 0.0 0.0 0.03693648 #Ba
0.00000000 4.03331143 0.00000000 0.0 0.0 0.03693648 #Ba
0.00000000 0.00000000 4.03331143 0.0 0.0 0.03693648 #Ba
4.03331143 0.00000000 4.03331143 0.0 0.0 0.03693648 #Ba
4.03331143 4.03331143 4.03331143 0.0 0.0 0.03693648 #Ba
0.00000000 4.03331143 4.03331143 0.0 0.0 0.03693648 #Ba
%endblock local_mode.mode_definition
#=========================================#
使用示例的3个输入文件,运行PASP可得到100组随机构型的输出文件: *.vasp 为结构信息(可作为VASP的POSCAR使用); *.ene_symbol 提供各构型对各种相互作用的贡献信息(平均到每个原胞,参见2.2.1.1节 distortion.ene_symbol 文件介绍); *.str_mode_amp 提供各局域模式大小信息,以下将举一例解释该文件含义。例如测试运行时, seed.in 文件内容为“ 644585526 151130088”(可用于复现随机结果),得到的1.str_mode_amp内容为:
#=========================================#
#undistorted structure #以下提供超胞的原始结构信息(所有局域模式为零)
15.243706845086 0.000000000000 0.000000000000 #基矢1
0.000000000000 15.243706845086 0.000000000000 #基矢2
0.000000000000 0.000000000000 15.243706845086 #基矢3
#以下各行:原子编号;元素编号;分数坐标(超胞基矢)
1 1 0.249999999602 0.249999999602 0.000000000000
2 1 0.249999999602 0.000000000000 0.249999999602
3 1 0.000000000000 0.249999999602 0.249999999602
4 2 0.249999999602 0.249999999602 0.249999999602
5 3 0.000000000000 0.000000000000 0.000000000000
6 1 0.749999999602 0.249999999602 0.000000000000
7 1 0.749999999602 0.000000000000 0.249999999602
8 1 0.500000000000 0.249999999602 0.249999999602
9 2 0.749999999602 0.249999999602 0.249999999602
10 3 0.500000000000 0.000000000000 0.000000000000
11 1 0.249999999602 0.749999999602 0.000000000000
12 1 0.249999999602 0.500000000000 0.249999999602
13 1 0.000000000000 0.749999999602 0.249999999602
14 2 0.249999999602 0.749999999602 0.249999999602
15 3 0.000000000000 0.500000000000 0.000000000000
16 1 0.749999999602 0.749999999602 0.000000000000
17 1 0.749999999602 0.500000000000 0.249999999602
18 1 0.500000000000 0.749999999602 0.249999999602
19 2 0.749999999602 0.749999999602 0.249999999602
20 3 0.500000000000 0.500000000000 0.000000000000
21 1 0.249999999602 0.249999999602 0.500000000000
22 1 0.249999999602 0.000000000000 0.749999999602
23 1 0.000000000000 0.249999999602 0.749999999602
24 2 0.249999999602 0.249999999602 0.749999999602
25 3 0.000000000000 0.000000000000 0.500000000000
26 1 0.749999999602 0.249999999602 0.500000000000
27 1 0.749999999602 0.000000000000 0.749999999602
28 1 0.500000000000 0.249999999602 0.749999999602
29 2 0.749999999602 0.249999999602 0.749999999602
30 3 0.500000000000 0.000000000000 0.500000000000
31 1 0.249999999602 0.749999999602 0.500000000000
32 1 0.249999999602 0.500000000000 0.749999999602
33 1 0.000000000000 0.749999999602 0.749999999602
34 2 0.249999999602 0.749999999602 0.749999999602
35 3 0.000000000000 0.500000000000 0.500000000000
36 1 0.749999999602 0.749999999602 0.500000000000
37 1 0.749999999602 0.500000000000 0.749999999602
38 1 0.500000000000 0.749999999602 0.749999999602
39 2 0.749999999602 0.749999999602 0.749999999602
40 3 0.500000000000 0.500000000000 0.500000000000
8 #nsite #共8个有局域模式的(中心)原子
1 4 1 3 #isite,isite2ia(isite),uc_site(isite),nmode
#第1个有局域模式的中心原子
#该原子在上述超胞中的编号为4
#该原子是原胞中第1个有局域模式的(中心)原子(以在local_mode-*.xsf中的出现顺序为准)
#该原子相关的局域模式数共3个(对应地,以下应列出3行数据)
1 1 0.081453051445
#该原子的第1种局域模式
#对应于local_mode-1.xsf的模式
#该模式的大小
2 2 0.142100422264
3 3 0.191733544595
2 9 1 3 #isite,isite2ia(isite),uc_site(isite),nmode
1 1 0.034895461810
2 2 -0.084312895260
3 3 -0.240252037953
3 14 1 3 #isite,isite2ia(isite),uc_site(isite),nmode
1 1 0.222075675504
2 2 -0.167235113016
3 3 0.138588431349
4 19 1 3 #isite,isite2ia(isite),uc_site(isite),nmode
1 1 -0.223240429075
2 2 0.187046512763
3 3 -0.214230248144
5 24 1 3 #isite,isite2ia(isite),uc_site(isite),nmode
1 1 0.263319144138
2 2 -0.148031542190
3 3 -0.267702750924
6 29 1 3 #isite,isite2ia(isite),uc_site(isite),nmode
1 1 0.107717267160
2 2 -0.244044246481
3 3 -0.200558481092
7 34 1 3 #isite,isite2ia(isite),uc_site(isite),nmode
1 1 -0.081994077120
2 2 0.043118831171
3 3 0.074635582067
8 39 1 3 #isite,isite2ia(isite),uc_site(isite),nmode
1 1 -0.263113832288
2 2 0.164899246181
3 3 0.057835567382
-0.007718100274 0.015755883075 0.011762573232 -0.003892679096 -0.003683198494 0.024630625555 #strain
#最后给出六种应变模式的大小
#=========================================#
另外,提供各不等价相互作用信息的 all_interaction_type_Spin_xyzform.dat 文件部分内容示例如下:
#=========================================#
icluster, #sites, which-cluster: 1 0 1
1 1 e_1 +e_2 +e_3
2 2 e_1^2 +e_2^2 +e_3^2
3 2 e_1 e_2 +e_1 e_3 +e_2 e_3
4 2 e_4^2 +e_5^2 +e_6^2
icluster, #sites, which-cluster: 2 1 1
5 2 u_{1,x}^2 +u_{1,y}^2 +u_{1,z}^2
6 3 u_{1,x}^2 e_1 +u_{1,x}^2 e_2 +u_{1,y}^2 e_2 +u_{1,y}^2 e_3 +u_{1,z}^2 e_1 +u_{1,z}^2 e_3
#=========================================#
其中第一行三个数字分别表示:第1种团簇(cluster),该团簇包含0个原子(即纯应变项),在纯应变项中编号为1。第二行两个数字分别表示:第1种相互作用(与distortion.ene_symbol中的第几种一致),阶数为1。第二行后面给出了相互作用的数学形式(展开式,未作合并同类项等处理)。在各行的相互作用展开式中,e_1表示第1种应变模式,u_{1,x}表示(代表簇中的)1号原子的x方向的局域模式,以此类推。
代表簇的坐标信息等仍在 local_mode_PM.dat 文件中提供,该文件内容含义参见2.2.1.1节。
备注
最后补充强调一点,局域声子模式中的原子位移与晶格应变操作是不可对易的。在PASP软件内部是先完成应变操作再进行原子位移的。当模型涉及应变与局域声子模式的相互作用项时,为保证模型的合理性与准确性,需要满足局域声子模式导致的原子位移与晶格应变操作对易性所导致的差异是高阶小量而可以忽略不计,这要求原子位移和晶格应变都足够小。
2.2.2 确定哈密顿量参数的机器学习方法 (Model Fitting_one_order)¶
在2.2.1节中提到,当我们采用各自由度的泰勒展开形式并适当淘汰、合并项之后,有效哈密顿量模型可以抽象成如下线性组合的形式:
其中 \(\left\{h_{j}\right\}\) 为线性组合的基函数(basis function),这部分是构型的函数; \(\left\{C_{j}\right\}\) 为线性组合的待定系数,这是与构型无关的相互作用参数。有效哈密顿量中不重要的常数项对应于 \(h_{1}\equiv 1\) 。
当我们计算多组构型的数据时,用上标 \(^{\left(i\right)}\) 表示构型的编号,则可以利用PASP以及第一性原理计算,得到多组 \(\left\{\left\{h_{j}^{\left(i\right)}\right\},H_{eff}^{\left(i\right)}\right\}\) 数据(获取方法详见2.2.1节)。本节将介绍如何利用这些数据确定哈密顿量参数 \(\left\{C_{j}\right\}\) 。
如果直接利用 \(\left\{\left\{h_{j}^{\left(i\right)}\right\},H_{eff}^{\left(i\right)}\right\}\) 数据,用最小二乘法拟合得到 \(\left\{C_{j}\right\}\) ,看似是很容易的。但是这样做有很多弊端。第一,拟合要求构型数大于待定参数个数,当待定参数有几千或几万个时,对于数据量的要求是很高的,这有时是不现实的。第二,若采用梯度下降、共轭梯度下降、拟牛顿法等方法最小化残差平方和(即最小二乘法),最终模型几乎会保留所有项,导致模型过于复杂,物理图像不够清晰,也不利于后续蒙特卡洛模拟或分子动力学模拟的计算效率。第三,当待定参数个数很多时,很容易引起过拟合问题,也就是说,拟合时看似精度很高,但对新构型能量的预测能力显著更差。
我们采用的思路是,先筛选出公式 (14) 中重要项,把不重要项的系数取成0,再拟合确定重要项的待定参数。也就是说,假定经过筛选共有个 \(p\) 重要项( \(p\) 也是待定参数个数),对公式 (14) 各项的脚标重新编号后可以写成如下形式:
这时再拟合就只需求解 \(p\) 个待定参数。这种筛选重要项的问题在计算机算法领域称为变量筛选(variable selection)问题,经典的算法有序列前向选择(sequential forward selection, SFS) 14 算法等。SFS算法是在初始模型中不含任何项的基础上,每次尝试在模型中增加一项,每一轮增加项都是挑选出能使拟合效果最好的项加到模型中。PASP支持使用改进版本的SFS算法,这里所谓的改进是指引入了测试集以减小过拟合风险。流程图如图2.2.1所示(图中 \(\widehat{\sigma^{2}}\) 为模型在训练集上的拟合方差, \(\widehat{\sigma{}'^{2}}\) 为模型在测试集上的平均预测方差, \(p_{C}\) 是参数个数上限)。

图2.2.1 使用了测试集的SFS(序列前向选择)算法流程图¶
我们设计了一套机器学习算法来高效可靠地处理我们遇到的这种变量筛选问题,我们称之为MLMCH方法(全称为Machine Learning Method for Constructing realistic effective Hamiltonian) 3 。这里所谓的机器学习方法与神经网络无关,因为神经网络所得的模型的简洁性和可解释性不足,而我们希望得到的是简洁、物理图像清晰的模型。我们之所以称之为机器学习方法,一是因为我们所处理的问题本身属于监督学习中的回归问题(即构型能量作为标记信息的问题);二是我们采用了机器学习中常用的技巧,如利用训练集确定模型参数后在测试集上检验模型的预测能力(以避免过拟合),采用了类似于 \(L_{0}\) 正则化项的技巧控制参数个数(避免参数过多,以保证模型的简洁性);三是在尝试在模型中删除、增加或替换某个项的过程中,会根据模型的拟合效果的变化来“学习”该项的“重要性”,从而加速后续的搜索过程。
传统的SFS方法只考虑在模型中不断增加项,因此一旦引入了不必要的项就无法再删去。我们为了弥补这个缺陷,在我们的MLMCH方法中还考虑了删除、替换项的可能性。虽然这样做增加了搜索的复杂度,但是我们通过改良算法,大幅提高搜索效率,使得我们的算法在即使考虑了删除、替换项之后仍然具有与SFS接近的效率(甚至在部分测试中具有更高的效率)。我们的算法在仅使用训练集和测试集时称为Method 1,在额外引入验证集时称为Method 2(根据我们的测试经验,Method 1的表现通常优于Method 2,所以我们最常用的方法是Method 1和Method 3)。训练集、验证集、测试集三者的简单对比如表2.2.1所示;Method 1的流程图如图2.2.2所示。图2.2.2中的 \(\left\{x_{j}^{\left(i\right)},y^{\left(i\right)}\right\}\) 对应于公式 (14) 的 \(\left\{h_{j}^{\left(i\right)},H_{eff}^{\left(i\right)}\right\}\) ,上标代表构型的编号;图中最后一行提到的SBS(sequential backward selection,序列后向选择)搜索是指依次尝试在模型中减少一个参数,每次选出其中拟合效果最佳的方案。SBS搜索步骤的主要目的是对某个给定模型的各个参数进行重要性排序,同时也为简化模型提供参考。SBS步骤的示意图如3.4.3所示。以Method 1为例,图2.2.2中“发现更优的模型?”是以 \(\left(\widehat{\sigma^{2}}\cdot \lambda^{p}\right)\) 更小作为模型更优的判据的(这与采用 \(L_{0}\) 正则化项时以 \(\widehat{\sigma^{2}}+\lambda^{p}\) 更小作为模型更优的判据类似,之所以改成乘 \(\lambda^{p}\) 是为了更方便设置 \(\lambda\) 值)。其中 \(\widehat{\sigma^{2}}\) 是指模型在训练集上的拟合方差, \(\lambda\ge 1\) 是人为设定的参数(该值越大,则越倾向于选择参数较少的模型)。在我们的算法中,会依次使用若干个递减的 \(\lambda\) 值,对每个不同的 \(\lambda\) 值分别搜索当前判据 \(\left(\widehat{\sigma^{2}}\cdot \lambda^{p}\right)\) 下的最优模型。再在这些“最优模型”中挑出 \(\widehat{\sigma{}'^{2}}\) (测试集上的预测方差)最小的模型,再在此基础上做一次SBS搜索(参数个数递减至1)。在此过程中会遇到多个值得推荐的模型,它们的详细信息都会在输出文件中给出,输出文件还会给出两个最推荐的模型,分别是预测效果最好的模型,和“在过拟合风险足够低的前提下”预测效果最好的模型。关于如果选择确定最终用于后续MC或MD模拟的模型,后文还会再详细讨论。
表2.2.1 拟合问题中训练集、验证集、测试集三者的异同点比较
数据集 |
训练集 |
验证集 |
测试集 |
|---|---|---|---|
是否必需 |
是 |
否 |
建议使用 |
可用于拟合参数 |
是 |
否 |
否 |
用于避免过拟合 |
否 |
是 |
是 |
在模型搜索过程中是否可用 |
是 |
是 |
否(最终测试用) |
样本量 |
\(n_{1}\) |
\(n_{2}\) |
\({n}'\) |

图2.2.2 MLMCH算法流程图¶
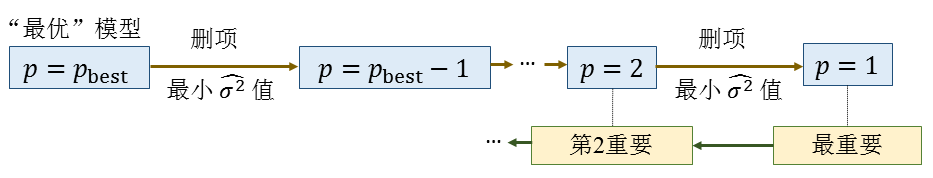
图2.2.3 通过SBS搜索对给定模型中的参数进行重要性排序的算法示意图¶
2.2.2.1 Method 1(默认形式的MLMCH方法)¶
默认形式的MLMCH方法所需的全部示例输入文件在 “example_public/Fitting_one_order/input” 文件夹下,包括 Fitting_Data_for_prediction.txt 、 Fitting_Dataset.txt 、 Fitting_Settings.txt 、 Fitting_Set_Lambda.txt 和 PASP.input 共5个输入文件。
备注
PASP.input 中只需写两行,一行选定模型“Model Fitting_one_order”,另一行写“%include cell.str”。注意我们无需提供 cell.str ,只需要通过“%include cell.str”使软件自动生成的默认 \(cell.str\) 文件生效即可(否则软件会因缺失部分设置而无法运行)。该Model实际上用不到 cell.str 中的任何设置,因此我们也可以自己提供任意一个格式正确的 cell.str 文件,这并不会影响结果。
Fitting_Dataset.txt (训练集数据)和 Fitting_Data_for_prediction.txt (测试集数据)分别需提供 \(n\) 行和 \({n}'\) 行数据, \(n\) 是训练集的构型数, \({n}'\) 是测试集的构型数。每行内容对应一种构型的数据。每行前面的若干数据是该构型对各种相互作用的贡献信息 \(\left\{h_{j}^{\left(i\right)}\right\}\) (平均到每个原胞),这些信息可在distortion.ene_symbol或*.ene_symbol中获取,例如根据2.2.1.1节中的distortion.ene_symbol内容,该行前面的部分可写成“ 0.243841635894 1.379560781428 0.550369143339 …… -1.060482829187”共计346个数据(有几种不等价相互作用就应该有几个数据);此外每行最后要补充一个数据,即第一性原理算出的 \(H_{eff}^{\left(i\right)}\) (一般是该构型能量减去相同超胞的参考态能量,再除以原胞数,取eV单位)。因此每行的数据个数为“不等价相互作用数+1”。这两个文件实际上是提供了 \(\left\{h_{j}^{\left(i\right)},H_{eff}^{\left(i\right)}\right\}\) 构成的矩阵。
软件并非以只读方式打开 Fitting_Dataset.txt 和 Fitting_Data_for_prediction.txt 两个文件,恰恰相反,PASP运行过程中经常改写这两个文件。所以请注意务必在运行前对这两个文件的初始版本做好备份。如需在相同条件下重新测试运行,请从备份的版本重新拷贝一份作为输入文件,不要直接使用被修改过的这两个文件(直接使用容易导致推荐模型的相互作用编号错乱或参数值错误)。我们这里修改数据文件内容的本意是为了在反复调用该模块时节省时间成本,但这需要非常细心严格地调整设置项以及需要后续数据处理,此外这样做实际上节省的时间并不多,因此为了稳妥起见,我们强烈建议只使用未被软件改动过的训练集、测试集数据文件作为输入文件。
示例文件中的 Fitting_Settings.txt 全部采用的是缺省设置,换言之,只需要默认版本的MLMCH方法时,可以省略该输入文件。注意在该文件中,仅在每行读取开头的数据(除了在第三行需要读取4个数据外),而后面的文字部分仅用于注释;该文件是逐行读取相应设置内容的,不允许任意调换两行顺序或增减行数等。决定使用Method 1的两项设置是“
1 !! SEARCH_TYPE”和“0 !! I_CHECK_SELFPREDICT”,前者决定了使用MLMCH方法,后者决定了不使用验证集。示例文件中的 Fitting_Set_Lambda.txt 全部采用的是缺省设置,换言之,只需要默认版本的MLMCH方法时,可以省略该输入文件。该输入文件中第一行提供两个数据,分别表示后续提供几行数据(通常每行数据对应一个新的值,对应一轮大的搜索循环),以及从其中第几行开始使用。后面每行有三个有效数据,分别是第几行数据、本轮循环中允许模型中包含的最大参数个数、本轮循环中采用的值。这里的示例文件中共提供20行数据,对应20个递减的值,其中最后一个值恰好为1.0。注意值取1.0的最后一轮循环可能会显著偏慢,如果希望节省搜索时间,可以考虑把第一行的第一个数据由20修改为19或者更小,从而回避值取1.0的较慢的搜索过程。
本模块暂不支持并行。
输出文件 Fitting_lambda_log.txt 给出不同 \(\lambda\) 值下搜索得到的最优模型的拟合预测效果信息。各列数据的含义依次是:第几个 \(\lambda\) 值; \(\lambda\) 值;允许的参数个数上限;该 \(\lambda\) 值对应的最优模型的参数个数;模型的拟合标准差;预测值的平均不确定度;平均预测误差(这里实际是平均预测方差的算术平方根,下同);模型的拟合标准差(梯度下降优化后);预测值的平均不确定度(梯度下降优化后);平均预测误差(梯度下降优化后)。最重要的两列是第4列(模型的参数个数)和最后一列(经梯度下降优化的平均预测误差)。一般设定的 \(\lambda\) 值是递减的,这导致第4列(参数个数)一般是递增的(也可能保持不变);而最后一列(预测误差)通常先降后升,其中下降阶段是正常的(随参数个数增加,预测效果变好,其中各模型都可以被看作是最优模型),而上升阶段是过拟合的(随参数个数增加,预测效果反而变差,说明可能引入了冗余的参数)。各个 \(\lambda\) 值对应的不同参数个数的模型的详细信息将输出到名为“Report_fitting_function_MID_GRAD_%d* ”(%d是一个整数,表示模型的参数个数;* 表示任意内容,包括“ .txt ”、“ _data.txt ”、“ _data2.txt ”、“ _LM.txt ”共4种情形;本节后面出现“ %d* ”时,含义都与此相同)的系列文件中。而最后一列(预测误差)先降后升的拐点(极小值点)对应的模型,是具有较低过拟合风险的推荐模型,相应信息将输出到名为“Report_fitting_best_safe_function_GRAD_%d*”的一组文件中,该模型的预测误差可在“ Report_fitting_best_safe_function_GRAD_%d.txt ”的最后一行查到(例如“Average prediction error= 3.131095897621887E-003”)。
输出文件 Report_fitting.txt 给出最后的SBS搜索过程中不同参数个数的模型的拟合预测效果信息。各列数据的含义依次是:模型的参数个数;模型的拟合标准差;预测值的平均不确定度;平均预测误差(这里实际是平均预测方差的算术平方根,下同);模型的拟合标准差(梯度下降优化后);预测值的平均不确定度(梯度下降优化后);平均预测误差(梯度下降优化后)。最重要的两列是第1列(模型的参数个数)和最后一列(经梯度下降优化的平均预测误差)。在SBS搜索过程中,不同参数个数的模型的详细信息将输出到名为“ Report_fitting_function_GRAD_%d* ”的系列文件中。在这些模型加上上一段提到的 Fitting_lambda_log.txt 中涉及的模型中,选出平均预测误差最小的模型,相应的信息输出到名为“Report_fitting_best_predict_function_GRAD_%d*” 的一组文件中;该推荐模型的预测误差可在“ Report_fitting_best_safe_function_GRAD_%d.txt ”的最后一行直接查到(“Average prediction error”信息)。
通常来说,我们倾向于推荐“ Report_fitting_best_safe_function_GRAD_%d* ”所对应的模型(具有较低过拟合风险的推荐模型);当更看重预测效果而忽略过拟合风险时,也可选用“Report_fitting_function_GRAD_%d*”对应的模型(平均预测误差最小的模型)。此外,根据实际需要,可能还会选用参数个数更少的模型,这时需要在 Fitting_lambda_log.txt 和 Report_fitting.txt 两个文件中,根据各模型的参数个数和预测误差来选择。需要注意的是,为了降低计算量,对于最优模型的搜索是不完备的,也就是说,搜索给出的最优模型可能是局部最优而非全局最优。偶尔Method 3(见2.2.2.3节)也会给出表现优于Method 1的模型,所以我们一般建议Method 3和Method 1都做一遍,再在其中(依据参数个数和预测误差)选出最合适的模型。
前面三段多次提到含有星号的文件,其中星号包括“ .txt ”、“ _data.txt ”、“ _data2.txt ”、“ _LM.txt ”共4种情形,下面依次介绍这四种文件;这里将它们分别记为“*%d.txt”、“ *%d _data.txt ”、“ *%d _data2.txt”、“ *%d _LM.txt”(这里的 *表示任意内容,%d为整数,表示模型的参数个数)。
输出文件 “*%d.txt”提供模型中各参数的原编号、拟合值以及不确定度等信息。示例如下:
#=========================================#
5 (Number of terms) #模型中参数个数(这里有5个参数)
1 0 #第1个参数:原0号参数(即常数项)
2 1 #第2个参数:原1号参数
3 3
4 5
5 15 #至此为止,这部分内容可用作Fitting_Set_Init.txt内容
===============================================
INFORMATION OF TERMS:
XM(1,1)=1;
XM(2,1)=2;
XM(3,1)=4;
XM(4,1)=6;
XM(5,1)=16;
B( 1 )= -2.017205356211482E-002 +- 3.325125338423869E-005
#第1个参数,拟合值及不确定度约为-0.02017+-0.00003,以此类推
B( 2 )= 1.772080263938097E-003 +- 7.546948702774408E-005
B( 3 )= -8.077197635063232E-003 +- 8.533043259079341E-005
B( 4 )= 1.859024576895017E-003 +- 4.260197138682767E-005
B( 5 )= 1.540675214074116E-003 +- 1.916551545772804E-005
Np= 5 Sigma_square= 6.960348094825395E-007 #参数个数;拟合方差
Sigma= 8.342870066605014E-004 #拟合标准差
Average prediction error= 8.123935535626463E-004
#平均预测误差(新版本添加)
#=========================================#
输出文件 “*%d_LM.txt”提供模型中除常数项以外的参数编号及拟合值,该文件可用作后续蒙特卡洛模拟的 J_best.dat 文件内容(这里的LM是Latticemodel的缩写,即PASP软件的旧称)。示例如下:
#=========================================#
287 4
#不等价相互作用数(与*.ene_symbol中的个数一致);保留参数个数
1 0.1772080263938097E-02 #保留原1号参数;1号参数值
3 -0.8077197635063232E-02 #保留原3号参数;3号参数值
5 0.1859024576895017E-02
15 0.1540675214074116E-02
#=========================================#
输出文件 “ *%d _data.txt ”和“ *%d _data2.txt ”分别给出训练集和测试集中各构型对应的实际能量和预测能量的比较,前者是训练集,后者是测试集。各列内容依次是构型编号(即原来第几行的数据)、实际能量(一般为第一性原理计算结果)、模型给出的预测能量、能量预测值的不确定度、预测偏差(预测能量减去实际能量)。
有时还会产生名为 “Data_Variables_Mapping_(1st Reducing)_%d.txt” (%d为整数,表示原始的不等价相互作用数)的输出文件。出现这样的输出文件时,意味着其中有些本该线性无关的相互作用的数据出现了明显线性相关的情形。遇到这种情况常见的原因包括但不限于以下情形:(1) 超胞过小或截断距离设置的过大,例如截断距离超过超胞最短边长的一半时,在周期性边界条件的影响下,较远距离相互作用的贡献可能会与较近距离相互作用的贡献合并,而无法分辨出各自的贡献。(2) 构型具有局限性,即如果不是完全随机采样,而是人为设置构型,有可能因为构型的局限性导致部分相互作用的贡献总是零。文件内容示例如下:
#=========================================#
1:1 #第1种不等价相互作用,对应于原来的第1种
2:2
……(省略若干行)
525:556
526:557 #第526种不等价相互作用,对应于原来的第557种
#=========================================#
可以看出,该文件列出了排除了明显冗余的相互作用项之后,实际有效的不等价相互作用个数,以及有效的不等价相互作用编号与原始编号的对应关系。这时,可以在 “Data_Special_Properties_%d.txt” (%d为整数,表示原始的不等价相互作用数)文件中查到其中冗余相互作用项的更具体的信息。
#=========================================#
24: Constant Variable
#原第24种相互作用贡献值为常数,即原第24列为常数列
59: Constant Variable
……(省略若干行)
29: Proportional to #16
#原第29种相互作用贡献值正比于第16种,即29、16列成正比
165: Proportional to #16
……(省略若干行)
Number of useless variables: 31
#因常数列或正比列而判定为冗余列的总数为31
#=========================================#
备注
注意这里为了简单与高效起见,仅初步筛查常数列和正比列这两种简单的线性相关情形。对于多列之间线性相关的复杂情形,这里没有进行细致检查。当遇到这些明显线性相关的情形时,建议逐个核对各个冗余项线性相关的实际原因(是因为超胞过小,还是因为构型具有局限性等等),根据经验和物理直觉判断这些项是否真的可以直接舍弃;如果认为不能直接舍弃,则应通过调整超胞大小或修改构型产生方式来避免线性相关的问题。
2.2.2.2 Method 2(使用验证集的MLMCH方法)¶
在Method 2中,会在Method 1的基础上附加一组验证集,在搜索最优模型过程中使用。当使用 Method 2时,Fitting_Dataset.txt (训练集数据)的 \(n\) 组数据会分成两部分,前面的 \(n_{1}\) 组作为训练集,后面的 \(n_{2}\) 组作为验证集。
为了选用Method 2,只需将2.2.2.1节示例文件中 Fitting_Settings.txt 文件(即“example_public/Fitting_one_order/input/Fitting_Settings.txt”,下同)的如下设置项修改为1:
1 !! I_CHECK_SELFPREDICT: Try 0 and 1. Usually 0 is better (but not always).
默认情况下,训练集和验证集各占一半。如需调整训练集和验证集的分配比例,只需修改Settings文件的如下设置项( \(n_{2}/n\) 的比值):
0.500E+00 !! MAX_Nncheck_BY_Nn: Usually 0.5 (Not important if I_CHECK_SELFPREDICT=0).
由于Method 2在绝大部分情形下表现不如Method 1,所以现已很少使用。
2.2.2.3 Method 3(使用测试集的SFS方法)¶
在Method 3中,采用传统的SFS方法,但引入了测试集以避免过拟合。在使用SFS方法逐个添加参数之后,会再做一组SBS搜索逐个减少参数。
为了选用Method 3,只需将2.2.2.1节示例文件中 Fitting_Settings.txt 文件的如下设置项修改为3:
3 !! SEARCH_TYPE: 1:My(Usually)! 2:add(complete)&delete. 3:only add(complete). 4:My(complete when adding). 5:My(Accurate add). 6:My(Accurate del)
输出文件 Fitting_lambda_log.txt 给出SFS添加参数过程中各模型的拟合预测效果信息。相应的各模型的详细信息将输出到名为“ Report_fitting_function_MID_GRAD_%d* ”的系列文件中。其中具有较低过拟合风险的推荐模型的相应信息将输出到名为“ Report_fitting_best_safe_function_GRAD_%d* ” 的一组文件中。输出文件 Report_fitting.txt 给出最后的SBS搜索过程中不同参数个数的模型的拟合预测效果信息。在SBS搜索过程中,不同参数个数的模型的详细信息将输出到名为“ Report_fitting_function_GRAD_%d* ”的系列文件中。在这些模型加上前述的 :guilabel:`Fitting_lambda_log.txt`中涉及的模型中,选出平均预测误差最小的模型,相应的信息输出到名为“Report_fitting_best_predict_function_GRAD_%d*” 的一组文件中。这些文件的内容解释参见2.2.2.1节。
2.2.2.4 自定义初始模型¶
如需自定义初始模型,只需将 Fitting_Settings.txt 文件(例如采用Method 1时,可基于”example_public/Fitting_one_order/input/Fitting_Settings.txt”文件修改;采用Method 2或3时,参见2.2.2.2节和2.2.2.3节内容修改后再作下述修改。下同)的如下设置项修改为5:
5 !! I_AUTOINIT: 1:Only constant term! 5: User-defined!(Use Fitting_Set_Init.txt) 2:Initially try each parameter to determine initial form.
默认的选项1是初始时只有常数项,而选项5是通过 Fitting_Set_Init.txt 文件自定义初始模型, Fitting_Set_Init.txt 的示例文件如下(#后面的内容为注释部分,无需写出;注意这里#号需要用空格或制表符与数据隔开;此外#号开头的首尾两行不算在文件内容之内):
#=========================================#
2 #第一行指出初始模型包含几个参数
1 0 #初始模型中的第1个参数 是编号为0的参数(即常数项)
2 1 #初始模型中的第2个参数 是编号为1的参数
#=========================================#
如果该文件内容空白,则初始时无任何项。
2.2.2.5 使用指定的模型拟合¶
如果不想进行最优模型搜索,而直接用指定的若干参数进行拟合,则需要在自定义初始模型(参见2.2.2.4节)的设置基础上,再将 Fitting_Settings.txt 文件的如下设置项修改为1:
1 !! I_ONLY_PREDICT: Usually 0. Use 1 when finally giving reports(with I_AUTOINIT=5).
该项设置为1的效果是,跳过搜索最优模型阶段,直接进入最后的逐个减参数的SBS搜索过程。在所有输出文件中,参数个数最多的模型对应的文件给出了指定模型的拟合结果。而通过参数个数递减的SBS搜索过程,可以给出指定模型中各参数的重要性排序(参见图2.2.3)。
2.2.2.6 强制保留部分参数¶
如需强制保留部分参数,需要将 Fitting_Settings.txt 文件的如下设置项修改为1:
1 ! I_HAVE_KEEP_TERM: Usually 0. When 1, use Fitting_Set_Term_Keep.txt
同时,需要按如下示例文件的格式编辑输入文件 Fitting_Set_Term_Keep.txt (#后面的内容为注释部分,无需写出;注意这里#号需要用空格或制表符与数据隔开;此外#号开头的首尾两行不算在文件内容之内):
#=========================================#
4 #共强制保留4个参数
1 0 #强制保留的第1个参数 是编号为0的参数(即常数项)
2 1 #强制保留的第2个参数 是编号为1的参数
3 2
4 5
#=========================================#
备注
如果 Fitting_Settings.txt 文件中 I_AUTOINIT 设置项取默认值1,则在初始模型中会自动添加所有的强制保留的参数;但如果 I_AUTOINIT 设置项取5,则需要人为在自定义时添加所有的强制保留的参数,而不会自动补充添加。如果某个参数设置为强制保留,但在初始时不在模型中,将按如下方式处理:在搜索最优模型过程中将该参数看作普通参数,根据表现决定是否加入模型中,但一旦符合要求而加入到模型中,该参数就不会再在减少参数或替换参数的步骤中被淘汰掉。
2.2.2.7 强制跳过部分参数¶
如需强制跳过部分参数,需要将 Fitting_Settings.txt 文件的如下设置项修改为1:
1 ! I_HAVE_SKIP_TERM: Usually 0. When 1, use Fitting_Set_Term_Skip.txt
同时,需要按如下示例文件的格式编辑输入文件 Fitting_Set_Term_Skip.txt (#后面的内容为注释部分,无需写出;注意这里#号需要用空格或制表符与数据隔开;此外#号开头的首尾两行不算在文件内容之内):
#=========================================#
2 #共强制跳过2个参数
1 0 #强制跳过的第1个参数 是编号为0的参数(即常数项)
2 50 #强制跳过的第2个参数 是编号为50的参数
#=========================================#
备注
如果 Fitting_Settings.txt 文件中 I_AUTOINIT 设置项取默认值1,则在初始模型中会自动避免添加强制跳过的参数,例如常数项;但如果 I_AUTOINIT 设置项取5,则需要人为在自定义时避免添加所有的强制跳过的参数。如果某个参数设置为强制跳过,但在初始时已包含在模型中,将按如下方式处理:在搜索最优模型过程中将该参数看作普通参数,根据表现决定是否从模型中淘汰,但一旦被从模型中淘汰,该参数就不会再在后续步骤中被添加回模型。
2.2.2.8 设定部分参数取值¶
如需设定部分参数取值,需要将 Fitting_Settings.txt 文件的如下设置项修改为1:
1 ! I_SET_TERM_VALUE: Usually 0. When 1, use Fitting_Set_Term_Value.txt
同时,需要按如下示例文件的格式编辑输入文件 Fitting_Set_Term_Value.txt (#后面的内容为注释部分,无需写出;注意这里#号需要用空格或制表符与数据隔开;此外#号开头的首尾两行不算在文件内容之内):
#=========================================#
3 #共设置3个参数值
1 0 0.0 #设置的第1个参数值;编号为0的参数(常数项);设置为0.0
2 1 3.5 #设置的第2个参数值;编号为1的参数;设置为3.5
3 4 2.2
#=========================================#
备注
当设定部分参数取值时,软件实际的做法是,先将所设定的参数值对能量的贡献从总能量中扣除,再按正常流程搜索重要相互作用项。需要注意的是,默认情形下,即使是已经设置参数值的参数,仍然可能在搜索过程中被选中保留,这种情况意味着软件认为人为设定的参数值不够准确,需要修正。如果在设定参数值之后,需要禁止软件再修正相应的参数值,则应再另外设置“强制跳过相应的参数”(参见2.2.2.7节内容)。
2.2.2.9 设定部分参数的线性约束关系¶
在模型公式 (14) 中,若已知部分相互作用参数 \(\left\{C_{j}\right\}\) 总是满足某种线性约束关系,如 \(C_{1}+C_{2}+C_{3}-1=0\) ,这意味着 \(C_{1}\) 、 \(C_{2}\) 、 \(C_{3}\) 中必有一个冗余参数,可以由另外两个参数代替。例如 \(C_{2}=-C_{1}-C_{3}+1\) ,代入公式 (14) 后可发现 \(C_{2}\) 的作用可完全由 \(C_{1}\) 、 \(C_{3}\) 代替。
需要设置这种参数之间的线性约束关系时,需要将 Fitting_Settings.txt 文件的如下设置项修改为1:
1 ! I_SET_TERM_DEPENDENT: Usually 0. When 1, use Fitting_Set_Term_Dependent.txt
同时,需要按如下示例文件的格式编辑输入文件 Fitting_Set_Term_Dependent.txt (#后面的内容为注释部分,无需写出;注意这里#号需要用空格或制表符与数据隔开;此外#号开头的首尾两行不算在文件内容之内):
#=========================================#
1 #共设置1个冗余参数(1个关系式)
1 2 3 #第1个冗余参数:2号参数,相应关系式右边有3项(之后需要3行信息)
0 1.0 #0表示常数项;1.0为约束关系式中常数项的值
1 -1.0 #1表示1号参数;-1.0为约束关系式中1号参数的系数
3 -1.0 #3表示3号参数;-1.0为约束关系式中3号参数的系数
#=========================================#
如果有多项约束表达式,按上述格式继续往后依次填写即可,两个表达式的信息之间不空行。
备注
使用该设置时,软件一定会改写 Fitting_Dataset.txt (训练集数据)和 Fitting_Data_for_prediction.txt (测试集数据)两个输入文件内容,请特别注意备份原版文件。
冗余参数的取值完全由其他参数值决定,因此不支持强制保留或跳过这些参数(参见2.2.2.6节、2.2.2.7节;设定强制保留、跳过时不起作用),不支持设定这些参数值(参见2.2.2.8节;设定参数值时不起作用),也不支持在自定义初始模型时添加这些参数(参见2.2.2.4节;只需添加其他的非冗余参数,而冗余参数会根据约束条件自动计算并添加;特别注意,如果在自定义初始模型时添加了冗余参数,会导致程序运行错误!)。例如,在示例文件中,2号参数设定为冗余参数后,它的取值完全由1号和3号参数决定;2号参数是否保留在模型中,取决于根据约束关系式算出的参数值是否为零。不支持强制保留、跳过2号参数,不支持设定2号参数值,也不允许在自定义初始模型时添加2号参数!
2.2.3 蒙特卡洛模拟与CG局域优化 (Model atomic_effective_H)¶
PASP中使用MC时,通常采用并行退火蒙特卡洛(parallel tempering Monte Carlo, PTMC)方法 11 12 13 ,参见2.1.3节介绍。
“Model atomic_effective_H”是适用于磁性、铁电、多铁等各种类型体系的通用的蒙特卡洛模拟模块。所需的输入文件可以比较容易地从“Model Invariant”阶段的输入文件修改得到。在2.2.1节中,我们曾分三种情况提供“Model Invariant”阶段的示例文件(即2.2.1.1节、2.2.1.2节和2.2.1.3节内容)。这里仅以2.2.1.3节内容(铁电体系 \(BaTiO_{3}\) )为例,介绍如何进行后续的蒙特卡洛模拟计算。而对于2.2.1.1节和2.2.1.2节的情形(磁性体系),也按类似的方式修改输入文件即可。
以铁电体系 \(BaTiO_{3}\) 为例,所需的全部输入文件在 example_public/atomic_effective_H/MC&CG_Ferroelectric_user_defined_mode_BaTiO3/input 文件夹下。本阶段所需的输入文件有6个: PASP.input 、 MC.input 、 cell.str 、 unit_cell.str 、 J_best.dat 和 local_mode_PM.dat 。
备注
PASP.input 内容与“Model Invariant”阶段(2.2.1节)基本一致,只需在最前面添加一行“%include MC.input”。另外由于已经在“Model Invariant”阶段获得 local_mode_PM.dat 文件,可将“
local_mode.read_interaction F”改为“local_mode.read_interaction T”,以直接从 local_mode_PM.dat 文件中读取不等价相互作用信息,节省计算时间。此外,需要根据蒙特卡洛模拟的需求,利用“block SuperCell”或 cell.str 文件扩胞;当需要计算热容等统计量时,需使用充分大的超胞(这里的充分大,可以通过逐渐增加扩胞数,查看统计量的收敛趋势判断),这通常大于“Model Invariant”阶段的超胞。扩胞数建议先取较小值测试,根据计算耗时和模拟所得物理量的收敛情况再酌情增加扩胞数。此外,初步测试时, MC.input 中 PT.nblk 、 nbeg_blk 、 nblk 不宜过大,否则测试时间可能很长。在初步测试顺利通过后,如果能初步判断模型是合理的(例如能够给出符合预期的基态;或者虽然MC给出的最低能量态不是基态,但根据有效哈密顿量模型,基态的能量确实更低,只是因为MC不够充分而没有找到基态),再适当增加计算量。在增加到足够大的计算量时,这三个设置的推荐值是4000、2000、500。
本模块支持并行,但须注意 MC.input 中PT.M必须是进程数的整数倍,否则软件会报错停止!
当我们使用 MC.input 文件时,习惯上在 MC.input 中提供实际的Model设置,即“Model atomic_effective_H”。为了让 MC.input 的“Model atomic_effective_H”设置生效(而 PASP.input 中的“Model Invariant”设置无效),最好将“%include MC.input”放在 PASP.input 的第一行(参见3.1.1节)。如果担心混淆,也可在 PASP.input 中将无效的
Model设置行注释掉。采用该Model时,“
local_mode.cal_para_method”设置项不起作用,所以与之相关的 distortion.pos 、 distortion.MAGMOM 等输入文件都不需要提供,以及相关的“local_mode.nstr_random”、“local_mode.generate_ordered_str”、 “block local_mode.mode_amp”等设置项也不起作用。涉及自旋自由度时,在“block Spin_values”中,磁性原子的设置值必须取2。原因是,在“Model Invariant”阶段中,软件对自旋矢量作归一化处理,所以所有的参数值都是基于归一化的自旋求出的,即采用2.1.1节各式中不带波浪号(tilde)的参数定义方式。
在“block local_mode.mode_definition”中,“15 lamp 0.0 2.76”也可以只写“15”。当加上“lamp 0.0 2.76”内容时,表示在大的模式下采用指数形式的单体势函数使能量快速上升(详见3.1.7.5节)。如果加上了“lamp 0.0 2.76”内容但暂时不使用指数形式的单体势函数,不要直接用#注释(因为在block内部,#不会被视为注释),建议在 PASP.input 或 MC.input 中添加行“
local_mode.non_polynomial_outer F”(缺省值为T)。如需给局域声子模式或应变自由度添加取值范围限制,或者其他约束条件,请参见3.1.5.6节内容。
J_best.dat 文件与2.2.2.1节输出文件“*%d_LM.txt”的格式一致,用于提供模型中实际采用的各参数编号及其拟合值;另参见3.7节介绍。
local_mode_PM.dat 可使用2.2.1节的同名输出文件。如果缺少这个文件,也可在 PASP.input 中设置“
local_mode.read_interaction F”,重新分析不等价相互作用(一般比较耗时,所以不建议反复计算)。MC.input (提供与蒙特卡洛模拟相关的设置,参见3.1.5节)中值得注意的常修改的设置项列举如下。
#=========================================#
Model atomic_effective_H #实际使用的Model
Read_init_spin F #是否读取初始磁矩信息。若T,读取amp_local_mode.in构型
atom_Heff.homogeneous_strain F #MC中是否开启strain自由度(均匀)
atom_Heff.inhomogeneous_strain F #MC中是否开启非均匀strain自由度
atom_Heff.relax_str T #是否CG优化
PT.M 64 #不同温度个数。注意必须是并行计算进程数的整数倍!
PT.low_temp 0.000001 #最低温,Hartree单位,0.00001约等于3.15 K
PT.high_temp 0.002 #最高温
PT.Tmesh 2 #温度值分布:1反比(1/T均匀),2正比(T均匀)
PT.nblk_T 400 # number of exchange steps
nbeg_blk 200 # number of exchange steps before measurement
nblk 50 # number of MC block steps. Default 10. 建议取10的整数倍
# 外磁场,沿z方向添加1 T磁场时设置为0 0 1(若自旋归一化,有效值为1/S)
#%block ExternalMagneticField
#0 0 1
#%endblock ExternalMagneticField
#=========================================#
输出文件 C_PTMC_K.dat 给出不同温度(即设定的若干副本的温度)下的热容统计值。第一列为温度(单位K),第二列为热容(某种约化后的单位,只有相对大小有意义)。如果热容呈先上升后下降的趋势,则拐点处温度为相变温度(误差导致的局域扰动不算)。
蒙卡所得最低能量态(不一定恰好找到体系的基态)信息由名为“lowest_ene.*”的一组文件提供。其中 lowest_ene.pos 给出POSCAR格式的超胞中各原子坐标信息; lowest_ene.ene_symbol 给出基态对于各种不等价相互作用的贡献值(仅 J_best.dat 文件中涉及的相互作用给出非零贡献值,其余取零值),其格式和含义与2.2.1节的distortion.ene_symbol基本相同(只是这里未考虑的相互作用项贡献取零);lowest_ene.str_mode_amp给出基态的各个局域模式以及应变的大小,其格式和含义参见2.2.1.3节的示例文件 1.str_mode_amp ; lowest_ene.amp_local_mode 以另一种更简洁的格式给出基态的各个局域模式以及应变的大小,示例如下:
#=========================================#
1 2.901785753659 -4.162823203431 3.369777246670
#第1个有局域模式的原子;该原子各局域模式的大小
2 3.186716707285 -4.515955600339 3.587299249456
3 4.424829790741 3.383234868544 4.547196266060
……(省略若干行)
512 3.469348208439 -3.276650367835 -3.992938680425
0.000000000000 0.000000000000 0.000000000000 0.000000000000 0.000000000000 0.000000000000 #strain
#=========================================#
该文件中,除了最后一行给出六个strain模式的大小外,其他每行分别提供超胞中各个具有局域模式的原子的编号、各局域模式大小。为了更清晰地理解该文件的内容,可以查看相应的 lowest_ene.str_mode_amp 内容,如下所示:
#=========================================#
#undistorted structure #以下提供超胞的原始结构信息(所有局域模式为零)
60.974827380343 0.000000000000 0.000000000000 #基矢1
0.000000000000 60.974827380343 0.000000000000 #基矢2
0.000000000000 0.000000000000 60.974827380343 #基矢3
#以下各行:原子编号;元素编号;分数坐标(超胞基矢)
1 1 0.062499999900 0.062499999900 0.000000000000
2 1 0.062499999900 0.000000000000 0.062499999900
3 1 0.000000000000 0.062499999900 0.062499999900
……(省略若干行)
2560 3 0.875000000000 0.875000000000 0.875000000000
512 #nsite #共512个有局域模式的(中心)原子
1 4 1 3
#isite,isite2ia(isite),uc_site(isite),nmode
#第1个有局域模式的中心原子
#该原子在上述超胞中的编号为4
#该原子是原胞中第1个有局域模式的(中心)原子(以在local_mode-*.xsf中的出现顺序为准)
#该原子相关的局域模式数共3个(对应地,以下应列出3行数据)
1 1 2.901785753659
#该原子的第1种局域模式
#对应于local_mode-1.xsf的模式;该模式的大小
2 2 -4.162823203431
3 3 3.369777246670
2 9 1 3 #isite,isite2ia(isite),uc_site(isite),nmode
1 1 3.186716707285
2 2 -4.515955600339
3 3 3.587299249456
3 14 1 3 #isite,isite2ia(isite),uc_site(isite),nmode
1 1 4.424829790741
2 2 3.383234868544
3 3 4.547196266060
……(省略若干行)
512 2559 1 3 #isite,isite2ia(isite),uc_site(isite),nmode
1 1 3.469348208439
2 2 -3.276650367835
3 3 -3.992938680425
0.000000000000 0.000000000000 0.000000000000 0.000000000000 0.000000000000 0.000000000000 #strain
#最后给出六种应变模式的大小
#=========================================#
以上 lowest_ene.str_mode_amp 文件中标为深红色加粗字体的内容,即为 lowest_ene.amp_local_mode 文件的内容。
最低能量态的能量取值(由模型估计,略去常数项,单位Hartree)可在 lowest_ene.ene 中查得。
还有一组后缀名为xsf的输出文件。这些文件可以直接拖到vesta软件查看(可以自动显示各个磁性原子的自旋矢量,或原子的位移矢量)。示例文件参见2.1.3节的 lowest_onlyspin.xsf 文件内容及其解释。对于不同的xsf文件,矢量部分可能是自旋矢量信息,也可能是局域铁电模式导致的原子位移矢量信息。这组文件有以下几种:
第一种,原子坐标取平衡位置坐标:
lowest_ene.global_lowest.originalpos.all_atoms_spin.xsf #全部原子的磁矩
lowest_ene.global_lowest.originalpos.onlyspin.xsf #磁性原子(非零磁矩)的磁矩
lowest_ene.global_lowest.originalpos.displacement_car.xsf #全部原子的位移(笛卡尔坐标)
lowest_ene.global_lowest.originalpos.displacement_dir.xsf #全部原子的位移(分数坐标)
第二种,原子坐标取实际坐标:
lowest_ene.global_lowest.actual.all_atoms_spin.xsf #全部原子的磁矩
lowest_ene.global_lowest.actual.displacement_car.xsf #全部原子的位移(笛卡尔坐标)
以上六种文件都是MC所得最低能量态的相关信息,当体系不考虑自旋模式或局域铁电模式时,会省略部分输出文件。若文件名中global替换为node*,表示相应编号的核遇到过的最低能量态信息。
如果还使用了CG优化步骤,则会生成加了前缀relax-的上述各个文件(包括各个xsf文件,以及前面提到的 lowest_ene.amp_local_mode 等文件),提供相应的CG优化之后的结构及自旋组态信息。
此外,如果不止关心最低能量态,还关心不同温度下的随机状态,可以查看T_*_final_( *部分为温度值,单位Hartree)开头的若干xsf文件,它们提供了各个温度副本的MC末态信息。同样包含了前面提到的六种文件(可能会省略部分文件)。
2.2.3.1 设置初始构型与计算指定构型的能量¶
如需自定义蒙特卡洛模拟的初始构型,需要在 MC.input 中将如下设置项修改为T:
Read_init_spin T #是否读取初始磁矩信息。若T,读取amp_local_mode.in构型
在本节所选Model中,通过 amp_local_mode.in 文件提供初始构型,即各个局域模式的大小以及各个strain模式的大小。该文件格式与输出文件 lowest_ene.amp_local_mode 一致(参见前文示例),需要依次提供超胞中各个具有局域模式的原子的编号、各局域模式大小。编号与原子坐标的对应关系,以及每个原子具有的局域模式数,可在 lowest_ene.str_mode_amp 文件中查得(如果一开始缺少该文件,可以先不设置初始构型,用很少的步数完成一次蒙特卡洛模拟,以获得该输出文件)。
另外注意,在“Model Invariant”分析指定构型或随机构型的 *.ene_symbol 文件时,也会同时产生相应的 *.amp_local_mode 文件,这种文件与此处需要的 amp_local_mode.in 文件格式是一致的,可以直接拿来使用(重命名即可)。
如果需要直接获取初始构型的能量(在 J_best.dat 设定的有效哈密顿量模型下的预测能量),可以在 MC.input 中将“ PT.nblk_T ”设置为0(即不进行蒙特卡洛模拟步骤),运行PASP时可以在标准输出(运行软件时可通过“> log.out”将标准输出的内容输出到 log.out 文件中)中找到如下相应内容:
Initial energy is: 0.15741225 Hartree 4.28344747 eV
其中依次给出了Hartree单位和eV单位的初态能量。
2.2.4 分子动力学模拟 (Model atomic_effective_H)¶
在PASP软件中,选择“ Model atomic_effective_H ”,并设置变量“ atom_Heff.MD T ”,即可进行分子动力学模拟。分子动力学模拟的主要思想是采用数值方法求解以下Newton方程和LLG方程:
其中, \(\vec{v}_{k}\) 和 \(\vec{m}_{i}\) 分别表示第k个原子的速度和第i个自旋的磁矩; \(\vec{F}_{k}\) 和 \(\vec{B}_{i}\) 分别表示第k个原子的受力和第i个自旋处的有效磁感应强度(由有效哈密顿量模型所决定); \(\vec{F}_{k}^{fl}\) 和 \(\vec{B}_{i}^{fl}\) 分别表示采用Langevin方法控温时的涨落力场和磁场。 \(\gamma_{L}\) 为等效旋磁比。 \(v\) 为Newton方程的阻尼系数; \(\alpha\) 为LLG方程的阻尼系数(Gilbert damping constant)。
目前,软件中主要采用velocity Verlet方法积分Newton方程,采用Mentink’s SIB method 或McLachlan’s SMP method积分LLG方程。关于如何选用不同的积分方法可以参见3.1.6节中的相关介绍。
以 \(MnFe_{2}O_{4}\) 为例,所需的全部输入文件在 example_public/atomic_effective_H/MD_MnFe2O4/input 文件夹下。本阶段所需的输入文件有7个: PASP.input 、 MD.input 、 cell.str 、 unit_cell.str 、 J_best.dat 、 local_mode_PM.dat 和 amp_local_mode.in 。
备注
对于动力学模拟,
Read_init_spin总应设为T,从而读取 amp_local_mode.in 文件中的信息作为初始结构进行模拟演化; amp_local_mode.in 文件格式规则参见2.2.3节(特别是2.2.3.1节)。本模块支持并行,但须注意文件中
block SuperCell扩胞的倍数必须是总进程数的整数倍,否则程序将报错。需要在“block atom_Heff.MASS_local_mode”中提供各局域模式的等效相对原子质量信息。提供的的质量数目即为局域模式的总数目(不计入应变模式,这与“Model Invariant”阶段产生的 local_mode-*.xsf 文件个数一致),顺序通常按照“block local_mode.mode_definition”中定义各局域模式时确定的先后顺序(如果实际局域模式数大于定义中写出的局域模式数,这时顺序应当与 local_mode-*.xsf 文件的编号一致)。局域模式仅涉及一个原子时,取元素的相对原子质量;涉及多个原子时,则需要使用相应的有效质量(例如局域声子模式,常以模式定义中各原子位移的平方为权重取相对原子质量的加权平均值)。
值得注意的重要设置项列举如下(主要涉及 PASP.input 和 MD.input 两个文件)。
#=========================================#
Model atomic_effective_H # 有效哈密顿量方法
atom_Heff.MD T # T表示打开分子动力学模拟
%block SuperCell
6 0 0
0 8 0
0 0 5
%endblock SuperCell
%block Z_values
25 26 8
%endblock Z_values
%block Spin_values
2 2 0
%endblock Spin_values
Read_init_spin T #是否读取初始磁矩信息。若T,读取amp_local_mode.in构型
local_mode.read_interaction T
Invariant.SOC F
# GSM.maxpower 1
# local_mode.GSM2lm_int T
fig.maxk 3
%block fig.mdist
1 11.0 6.7
%endblock fig.mdist
Invariant.power 3 # the max power of invariant
Invariant.strain_power 0 # the power of strain
Invariant.pure_strain_power 0 # the power of pure strain
%block local_mode.mode_definition
2 # method, 2 is the general one, as below
60 # num of modes
1 spin
1
4.246239888184 4.246239888184 4.246239888184 1.0 0.0 0.0
1 spin
1
4.246239888184 4.246239888184 4.246239888184 0.0 1.0 0.0
1 spin
1
4.246239888184 4.246239888184 4.246239888184 0.0 0.0 1.0
……(为节省篇幅,省略中间部分内容)
1
3
5.215531205855 5.215531205855 7.523188458697 1.0 0.0 0.0
1
3
5.215531205855 5.215531205855 7.523188458697 0.0 1.0 0.0
1
3
5.215531205855 5.215531205855 7.523188458697 0.0 0.0 1.0
%endblock local_mode.mode_definition
%block atom_Heff.MASS_local_mode
54.938 54.938 54.938 ……(省略中间部分数据) 15.999 15.999 15.999
%endblock atom_Heff.MASS_local_mode
MD.Delta_t 0.5 # fs
atom_Heff.MD_time 4.0d3 # fs
SLD.mode 2 # 1表示MD/2表示SD/3表示SLD
LLG.method 2 # 1表示RK4方法/2表示SIB方法/3表示SMP方法
LLG.damping 6.0d-3 # Alpha
MD.damping 4.0d-2 # Beta, unit: u/fs
MD.temperature 80.0 # unit: K
MD.aft_step 1
MD.int_step 20
MD.init_velocity_temp 0.0d0 # unit: K
MD.B_x 0.0d0 # unit: T
MD.B_y 0.0d0 # unit: T
MD.B_z 0.0d0 # unit: T
MD.output_amp_lm T # T表示输出中间过程
#=========================================#
动力学模拟的输出文件主要有4个: amp_local_mode.out 、 temperature-t.dat 、 S_tot-t.dat 和 energy-t.dat 。如果设置“ MD.output_amp_lm T ”,则程序将生成一个名为 amp_local_mode.mid 的目录,其中包含多个 amp_local_mode.* 文件,每个文件记录了当次观测时体系的构型信息。
输出文件 amp_local_mode.out 中包含体系在模拟结束时的构型信息,其格式与 amp_local_mode.in 文件完全一致,且目录 amp_local_mode.mid 下的每个文件的格式也与 amp_local_mode.in 文件完全一致。
输出文件 temperature-t.dat 记录了每次观测时体系的统计温度,示例如下:
#=========================================#
0.000000E+00 0.000000E+00
3.850483E+00 0.000000E+00
7.561680E+00 0.000000E+00
1.110774E+01 0.000000E+00
1.446748E+01 0.000000E+00
1.781564E+01 0.000000E+00
2.075857E+01 0.000000E+00
2.375723E+01 0.000000E+00
2.562883E+01 0.000000E+00
2.775630E+01 0.000000E+00
2.984838E+01 0.000000E+00
3.199036E+01 0.000000E+00
3.430230E+01 0.000000E+00
3.617140E+01 0.000000E+00
3.762378E+01 0.000000E+00
3.900152E+01 0.000000E+00
……(省略若干行)
#=========================================#
该文件中,左侧一列为自旋系统的温度,右侧一列为晶格系统的温度,单位统一为开尔文(K)。
输出文件 S_tot-t.dat 记录了每次观测时体系的总自旋,示例如下:
#=========================================#
0.000000E+00 0.000000E+00 -1.920000E+03 1.920000E+03
-2.023891E+00 3.544137E+00 -1.917516E+03 1.917520E+03
-2.166884E+00 3.832520E+00 -1.915198E+03 1.915203E+03
-5.882759E+00 6.226149E+00 -1.912897E+03 1.912916E+03
-3.319592E+00 9.451351E+00 -1.910345E+03 1.910371E+03
-7.213153E+00 1.174391E+00 -1.908071E+03 1.908085E+03
-3.860357E+00 -3.671226E-01 -1.905609E+03 1.905613E+03
-2.071153E+00 -1.640835E+00 -1.903417E+03 1.903418E+03
-3.753091E+00 -1.277366E+00 -1.900741E+03 1.900745E+03
-4.627319E+00 3.103064E+00 -1.897576E+03 1.897584E+03
-2.723718E+00 1.580270E+00 -1.894612E+03 1.894615E+03
2.186829E-01 3.012630E+00 -1.892574E+03 1.892576E+03
4.373851E-01 3.151187E+00 -1.889818E+03 1.889821E+03
-2.191931E+00 3.873266E+00 -1.886974E+03 1.886980E+03
1.348611E+00 8.513628E+00 -1.884661E+03 1.884681E+03
2.830424E+00 1.170689E+01 -1.881422E+03 1.881460E+03
……(省略若干行)
#=========================================#
该文件中,从左到右四列分别为体系总自旋的x方向分量、y方向分量、z方向分量和模长大小,单位统一为约化普朗克常数( \(\hbar\) )。
输出文件 energy-t.dat 记录了每次观测时体系的能量,示例如下:
#=========================================#
-2.389221E+01 0.000000E+00 -2.389221E+01
-2.383720E+01 0.000000E+00 -2.383720E+01
-2.378433E+01 0.000000E+00 -2.378433E+01
-2.373259E+01 0.000000E+00 -2.373259E+01
-2.368411E+01 0.000000E+00 -2.368411E+01
-2.363542E+01 0.000000E+00 -2.363542E+01
-2.359239E+01 0.000000E+00 -2.359239E+01
-2.354907E+01 0.000000E+00 -2.354907E+01
-2.351762E+01 0.000000E+00 -2.351762E+01
-2.348072E+01 0.000000E+00 -2.348072E+01
-2.344607E+01 0.000000E+00 -2.344607E+01
-2.341537E+01 0.000000E+00 -2.341537E+01
-2.337948E+01 0.000000E+00 -2.337948E+01
-2.334855E+01 0.000000E+00 -2.334855E+01
-2.332195E+01 0.000000E+00 -2.332195E+01
-2.329260E+01 0.000000E+00 -2.329260E+01
-2.326415E+01 0.000000E+00 -2.326415E+01
……(省略若干行)
#=========================================#
该文件中,从左到右三列分别为体系总的势能、动能和总能量,单位统一为哈特利(Hartree)。
除上述输出文件外,在PASP程序的标准输出中,还将包含体系的温度、能量、热容、平均磁化强度、磁化率等测量结果,示例如下:
#=========================================#
atom_Heff_MD_main done
Spin Temperature (Kelvin): 72.289
Lattice Temperature (Kelvin): 0.000
Avg. Energy per Site (eV): -0.045740
Specific Heat (k_B): 70.638472
Avg. Magnetization (2muB·S): 0.132470
Susceptibility (M^2/Hartree): 357.694190
MD used time (minute): 11.201
Lattice model simulation is finished gracefully.
#=========================================#
2.3 全局结构搜索方法¶
晶体全局结构搜索可以在已知晶体化学式和外部条件的前提下找到体系的晶体结构,是其他物性分析的基础。接下来以 \(BiFeO_{3}\) 为例来介绍全局结构搜索的方法。该体系空间群为R3c,基态为G型反铁磁。
首先介绍微扰空间群法寻找钙钛矿的基态。例如,我们知道钙钛矿材料 \(ABO_{3}\) 的基态(R3c空间群)是高对称性结构 \(Pm\bar{3}m\) 的子结构,空间群是 \(Pm\bar{3}m\) 的子群。那么,我们可以在生成初始结构的时候,给高对称性 \(Pm\bar{3}m\) 结构施加随机扰动,然后使用 \(Pm\bar{3}m\) 的子群对称性对微扰后的结构做对称化,这样就得到了具有子群对称性的初始结构了。同样,在使用遗传算法生成子代的时候,对畸变模式本身做遗传演化,即对畸变子结构跟高对称性结构的结构差异做遗传演化,会比传统的切割拼接方法更有效。
接下来,我们以 \(BiFeO_{3}\) 为例来介绍微扰空间群方法。首先我们不考虑磁性,只寻找基态结构。因此,在VASP的输入文件INCAR里,我们需要预先设定磁性,我们在 INCAR 中设定G-AFM。全局结构搜索需要的输入文件包括 PASP.input , cell.str , unit_cell.str , run_pvasp.sh (用于批量提交VASP作业)和若干VASP计算相关的输入文件,全部示例输入文件见 “example_public/BiFeO3/1.GA/input” 文件夹下。
备注
初始结构文件建议采用具有较高对称性的结构。
示例文件中 run_pvasp.sh 内容和用于提交任务的 pasp.slurm 、 vasp.slurm 文件内容与所用的集群相关。这里是以鸿之微内蒙A区为例, pasp.slurm 用于提交单次的PASP作业;在执行该PASP作业过程中,会调用 run_pvasp.sh 脚本,该脚本用于批量提交所有的VASP作业;在 run_pvasp.sh 脚本中,会建立多个文件夹,分别使用 vasp.slurm 脚本提交VASP作业;在本组示例文件的 vasp.slurm 中,连续调用了三次VASP,依次采用了更高的精度,实际上 vasp.slurm 调用VASP的次数以及精度等设置细节,可根据用户实际使用需求进行修改。另外特别注意,这三个文件中涉及程序路径和用户名(如 run_pvasp.sh 中的df102708)的设置,需要注意修改。
示例文件中三组INCAR_* 和KPOINTS_* 是依次采用更高精度进行弛豫计算,实际上也可仅用一组INCAR和KPOINTS完成弛豫计算(此时需要相应修改vasp.slurm内容)。
PASP.input 文件部分关键设置项如下所示。
#=========================================#
Model GA # 全局结构搜索模块
GA.population_size 10 # Default 0,每一代的种群大小
GA.max_generation 10 # Default 0,遗传算法迭代次数
save_str.nsave_str 20 # 最终输出的低能结构数量
GA.perturbation T # Default F,是否开启微扰方法
GA.Pher 0.85d0 # Default 0.85d0,遗传算法交叉的概率
GA.Pmut 0.15d0 # Default 0.10d0,遗传算法突变的概率
# Pperm=1.0d0-Pher-Pmut,置换的概率
GA.lchk_mut T # Default T, 检查突变后的结构,舍弃不合理结构
str.init_sym T # 结构初始对称性
GA.symmetrize_str T # Default F, T表示对结构做对称化
GA.parallel T # Default F,T表示使用并行方法
GA.useBCM T # Default T,判断结构相似性时,使用BCM方法
GA.tol_BCM 0.01d0 # Default 0.01d0,BCM方法的误差
GA.pressure 0.0d0 # Default 0.0d0 Gpa,外界压力大小
#=========================================#
在运行过程中,会产生若干POSCAR_* 作为VASP的输入文件 POSCAR 。主要的输出文件是 Saved_*.POS ( *部分为编号),个数与“ save_str.nsave_str ”的设定值一致,这些即是经搜索找到的低能结构(后续处理参见4.1节)。
在以上讨论中,我们使用微扰空间群方法来生成初始结构,并且对畸变模式做遗传算法演化。当我们不知道体系基本框架时,我们也可以使用随机空间群方法来生成初始结构,使用传统的切割拼接方法来演化算法生成子代。此时,我们需要关闭GA.perturbation,然后需要添加以下设置项,这样我们就可以使用随机空间群方法来全局结构搜索:
#=========================================#
GA.gen_ran_str_sym T # Default F,T表示使用随机空间群方法来生成初始结构,做全局结构搜索。生成空间群为GA.spg_front到GA.spg_rear的随机结构
GA.spg_front 1 # Default 1,起始空间群号
GA.spg_rear 230 # Default 230,终点空间群号
# 修改下面这一行为F
GA.perturbation F # Default F,是否开启微扰方法
#=========================================#
以上关于结构搜索的讨论中,我们没有讨论磁性自由度。当我们在PASP中考虑磁性自由度时,在INCAR中不需要预先设定磁性,此时我们需要在 PASP.input 中添加以下关于磁性的命令:
#=========================================#
GA.magnetic T # Default F
# 自旋值,Fe的自旋值为3左右
%block Spin_values
0 3 0
%endblock Spin_values
GA.random_magnetic F # Default T. If T, generate magnetic configuration randomly, if F, generate magnetic configuration according to the magnetic space group
GA.lAFM_GA T # Default F. If T, we only consider AFM state
GA.lmut_mag_GA T # Default F, whether mute the magnetic configuration
GA.flip_spin_rate 0.25d0 # Default 0.2d0, the possibility of moment mutation
GA.ldiff_str_with_mag_GA T # Default F, define a same structure with same configuration and same mag
GA.lread_OUTCAR_mag F # Default F, read the magnetic from the OUTCAR, update the magnetic configuration
#=========================================#
以上所有讨论中,晶格常数和原子位置都可以迭代优化。但当我们知道晶胞形状时,我们可以只进行原子位置的优化,而不改变晶胞形状。此时我们需要添加以下命令:
#=========================================#
GA.fixlattice F # Default F,固定lattice
GA.fix_cell F # Default T,固定cell
# when lfixlattice is T, lfix_cell is T
#=========================================#
当体系受到面内应力时,即面内晶格常数固定,面外晶格常数不固定时,可以加入以下命令行:
#=========================================#
GA.biaxial_strain T # Default F。If T, we apply in-plain strain
# 固定面内应力,面内晶格常数
%block GA.lat_fix_2D_Ang
5.5225 0.0 0.0
0.0 5.5225 0.0
%endblock GA.lat_fix_2D_Ang
#=========================================#
2.4 紧束缚模型方法¶
紧束缚模型(Tight Binding Model,TB)方法被广泛应用于能带分析、磁相互作用研究,磁电耦合等方面的研究中。其主要思想为将晶格格点附近的电子以原子束缚态的形式表示,借助原子轨道线性组合(LCAO)来表示共有化电子波函数,从而求解固体薛定谔方程。
PASP中TB模块必要的两个输入文件为: cell.str 和 PASP.input ,其中 cell.str 文件给出研究对象的结构信息, PASP.input 文件控制具体所采用的TB方法种类、模型内各参数大小、元素类别、磁性参数等相关信息。部分TB方法除了上述两输入文件外还需要其他额外文件,我们将在方法的具体介绍中给出。
2.4.1 自定义紧束缚模型¶
PASP的自定义紧束缚模型方法支持手动输入哈密顿量矩阵中的所有在位能参数及跃迁参数,该方法仅需知道组成紧束缚哈密顿量的轨道数,并给相应矩阵元赋值,即可得到费米能级,能带结构,态密度等信息。
自定义紧束缚模型方法输入文件:
在自定义紧束缚模型模块中,输入文件包括主输入文件 PASP.input 和结构文件 cell.str 。以石墨烯带为例,输入文件实例在 “example_public/TB/TB_user_defined/input” 文件夹下。
注意:
block SuperCell ”扩胞;Hop_Parameters_specified_using_site ”和“ Hop_Parameters_specified_using_dis ”必须一T一F;Z_values 应取每个元素所考虑轨道的电子数,而非元素的原子序数。#=========================================#
Model TB #使用TB模块
%block Orbitals #元素轨道数目及在位能信息,这里考虑石墨烯两带模型
1 1 #元素类型1,考虑轨道数
0 #在位能,石墨烯两带模型中设置为0
%endblock Orbitals
Different_hop_path 1 #考虑跃迁的路径数
Hop_Parameters_specified_using_site F #是否采用点位信息表示跃迁路径
Hop_Parameters_specified_using_dis T #是否采用距离信息表示跃迁路径
TB.complex_hop F #TB跃迁参数是否选用复数
%block Hop_Parameters #跃迁参数设置
1 1 2.67958 #跃迁的元素编号,此处为C-C跃迁;跃迁距离,等于或略大于键长
-2.9 #跃迁参数,“类型1轨道数×类型2轨道数”的矩阵
%endblock Hop_Parameters
%block kgrid_Monkhorst_Pack #K点网格设置
100 0 0 0
0 1 0 0
0 0 1 0
%endblock kgrid_Monkhorst_Pack
%block DensityOfStates
#态密度计算设置,最低能、最高能、展宽、能量区间内取点数以及单位(eV或Hartree)
0.00 0.00 0.200 1000 Hartree
%endblock DensityOfStates
%block Z_values #每个原子参与跃迁的电子数
1
%endblock Z_values
BandLinesScale ReciprocalLatticeVectors #计算能带设置
%block BandLines
1 0.000000000 0.000000000 0.000000000 \Gamma
1000 0.500000000 0.00000000 0.000000000 X
%endblock BandLines
#=========================================#
自定义紧束缚模型方法输出文件:
2.4.2 Slater-Koster紧束缚模型(S-K TB)¶
S-K TB方法简介
S-K TB方法由J. C. Slater和C. F. Koster于1954年提出 15 ,使用两中心积分方法,对紧束缚模型中的矩阵元参数进行了推导,PASP集成的S-K TB在原始的基础模型上加入了自旋轨道耦合(SOC)以及描述电子间库伦排斥作用的平均场近似形式下的Hubbard排斥作用 1 。各项具体形式为:
指标 \(i\) 表示不同位点, \(m\) 表示不同轨道, \(s\) 表示不同自旋,其中第一项为各轨道在位能 \(\left(\epsilon_{im}\right)\) 以及轨道之间跃迁 \(\left(t_{im,jn}\right)\) 的哈密顿量,第二项为SOC哈密顿量,第三项为Hubbard排斥作用哈密顿量,哈密顿量对应的波函数均为单电子波函数。
通过对模型内的各参数进行测试,可得到研究目标量随不同参数变化的变化情况,进而定性地从不同角度对问题进行分析。
S-K TB方法输入文件:
在S-K TB模块中,输入文件仍为 PASP.input 和 cell.str ,以下以 \(NiO_{6}\) 正八面体团簇为例,输入文件实例在 “example_public/TB/SKTB/input” 文件夹下。
备注
建议在 cell.str 提供超胞信息,不建议用“
block SuperCell”扩胞;“
Hop_Parameters_specified_using_site”和“Hop_Parameters_specified_using_dis”必须一个为T,另一个为F;在TB模块中,
Z_values应取每个元素所考虑轨道的电子数,而非元素的原子序数。在block “
Slater_Koster_Hop_Parameters”中,hopping积分项的具体含义可参考原始文献 15 ;在“
block Orbitals”中,原子在位能大小,对应了哈密顿量矩阵中的对角元,其数值可由DFT计算体系得到的DOS图中大致估计(主峰附近对应的能量值),轨道顺序为p轨道 \(\left(p_{x},p_{y},p_{z}\right)\) 、d轨道 \(\left(d_{z2-r2},d_{x2-y2},d_{yz},d_{zx},d_{xy}\right)\) 、f轨道 \(\left(f_{xyz},f_{x3},f_{z3},f_{x\left(y2-z2\right)},f_{y\left(z2-x2\right)},f_{z\left(x2-y2\right)}\right)\) 。在“
block Slater_Koster_Hop_Parameters”中, \(V_{pdσ}\) 等数值的选取可参考W.A.Harrison所著“Electronic structure and the properties of solids - The Physics of the Chemical Bond”一书或其他文献,具体见3.1.9节参数“block Slater_Koster_Hop_Parameters”相关说明。输入文件中的“
magnitude_SOC”为SOC强度系数,“TB.U”为Hubbard排斥作用系数,分别对应于公式 (18) 哈密顿量中的λ与U,单位为eV,通常λ比U小一个数量级。其中参数λ可以根据其表达式 \(\left(g_{s}-1\right)\frac{1}{2}\frac{1}{4\pi \varepsilon_{0}}\frac{Ze^{2}}{r^{3}}\frac{1}{m_{e}^{2}c^{2}}\) 进行大致估算;参数U值大小,通常与第一性原理计算中常用的U值一致即可。“
Sn_basis”项决定了自旋量子化轴的z轴方向,若设置为F,则其始终沿晶胞c轴方向,若设置为T,则其为自旋所指方向。这一差异会导致在展开哈密顿量时,SOC项的形式产生差异,进而使得哈密顿量矩阵形式产生差异。详见3.1.9节“Sn_basis”相关说明。模型中哈密顿量矩阵元的解析形式,可利用各轨道按球谐函数线性组合的形式,代入哈密顿量各项的表达式中求得。例如在 \(NiO_{6}\) 正八面体团簇示例中,
Sn_basis设置为F,Ni原子3d轨道的哈密顿量矩阵形式如下:
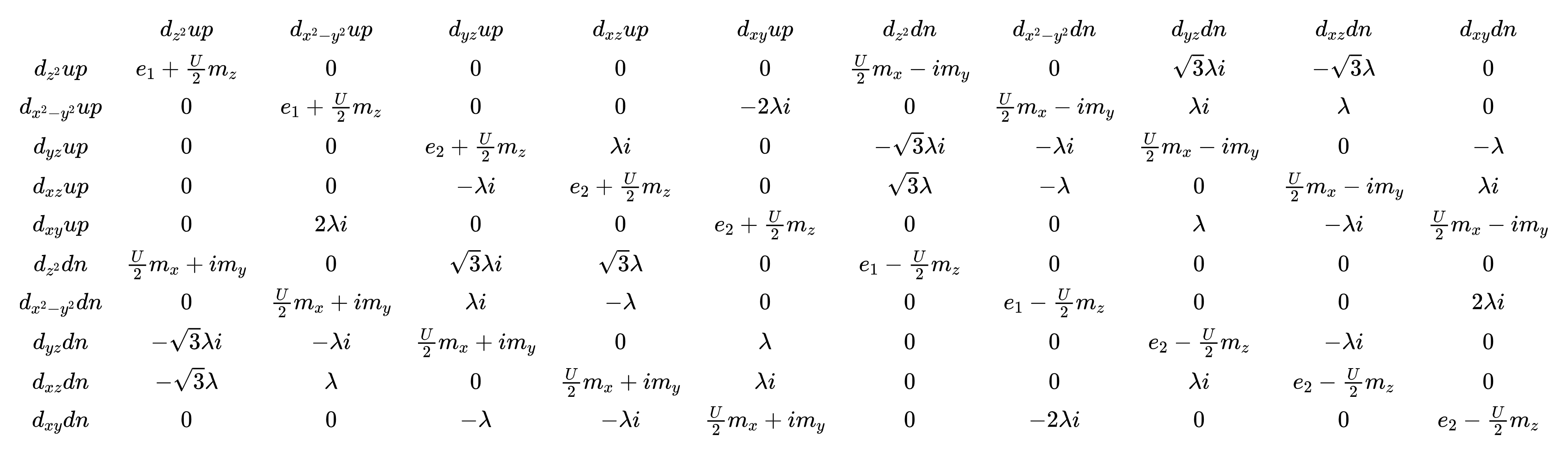
PASP.input 值得注意的若干设置项如下(参见3.1.9节的设置项说明)。
#=========================================#
Model TB #使用TB模块
USE_Slater_Koster_Model .TRUE. #若使用S-K TB,则设置为TRUE
use_rotation_matrix T #用旋转矩阵处理角动量,在使用S-K TB时此项必须设置为.TRUE.
#以下部分对应跃迁轨道信息及H_0部分
#===================== Orbital & H_0 ========================
Hop_Parameters_specified_using_site .FALSE.
Hop_Parameters_specified_using_dis .TRUE. #这两项决定采用位矢还是距离来表示跃迁路径
%block Z_values
8 6 #Ni的3d轨道有8个电子,O的2p轨道有6个电子
%endblock Z_values
#注意,在TB模块中,此处的Z_values指每个元素所考虑跃迁轨道的实际电子数,而非元素的原子序数。
overlap.distance 4 # 交叠距离,略大于原子间成键的长度即可。单位Bohr。
Different_hop_path 1 #考虑跃迁的壳层对的数目
%block Slater_Koster_Hop_Parameters
1 2 3.967974 # 元素类型1, 元素类型2, 键长(单位Bohr)
1 # 壳层对数目
3 2 2 1
# 元素1所考虑轨道的主量子数
# 元素1所考虑轨道的角量子数
# 元素2所考虑轨道的主量子数
# 元素2所考虑轨道的角量子数(本例对应3d、2p)
-2 3 # S-K TB对应的hopping积分项的数值(p轨道与p轨道间为V_ppσ V_ppπ, p轨道与d轨道间为V_pdσ V_pdπ,p轨道与f轨道间为V_pfσ V_pfπ)
%endblock Slater_Koster_Hop_Parameters
%block Orbitals #轨道及在位能信息(单位eV),可从DFT得到的DOS图大致判断
1 5 #元素1,所考虑轨道的数目(本例Ni 3d轨道,数目为5)
0.7 0.7 -0.2 -0.2 -0.2 #各轨道上的在位能大小 (本例为Eg(d_x2-y2,d_r2)轨道与T2g(d_xy,d_yz,d_zx)轨道的在位能)
2 3 #元素2,所考虑轨道的数目(本例O 2p轨道,数目为3)
-1.7 -1.7 -1.7 # 各轨道上的在位能大小(本例为2p(px,py,pz)轨道的在位能)
%endblock Orbitals
%block orbitals_nl #考虑跃迁的壳层信息
1 1 #第一种元素 考虑一个轨道
3 2 #3d轨道的n l
2 1 #第二种元素 考虑一个轨道
2 1 #2p轨道的n l
%endblock orbitals_nl
#===================== Orbital & H_0 ========================
#以下部分对应自旋、H_SOC以及H_Hubbard
#=============== ========= Spin ===========================
%block Spin_values
2 0 #元素1的磁矩,元素2的磁矩
%endblock Spin_values
spin_noncollinear T #是否开启非共线
Sn_basis F
# 若设置为F,自旋z量子化轴为晶胞z轴
# 若设置为T,自旋z量子化为与自旋方向共线的轴,这一项会使哈密顿量矩阵形式不同。
nspin_directions_set 1 #设定自旋方向的原子数目(本例为1个Ni原子)
%block spin_directions_set
1 -0.707107 0.707107 0.000000 #原子序数及其对应的自旋方向
%endblock spin_directions_set
#=============== ========= Spin ===========================
#=============== ========= H_SOC ===========================
nsites_SOC 1 #考虑SOC的原子数目(本例考虑1个Ni原子)
%block magnitude_SOC
1 0.3 #原子序数,对应的SOC强度λ(单位为eV)
%endblock magnitude_SOC
#=============== ========= H_SOC ===========================
#=============== ========= H_Hubbard ===========================
TB.U 2 #Hubbard排斥作用系数U大小,单位为eV
#=============== ========= H_Hubbard ===========================
#=========================================#
S-K TB方法输出文件
输出文件包括程序标准输出(可指定输出到自定义文件中,如 log.out );哈密顿量矩阵文件: H_2D.dat , H_I.dat , H_R.dat , H.dat , Hso.dat , Ht.dat , Htot.dat , HU.dat ;本征值文件: EIG_NSOC.dat , EIG.dat ;波函数文件: WF_I.dat , WF_NSOC.dat , WF_PROJ.dat , WF_R.dat , WF_SPIN.dat , WF.dat ;密度矩阵文件: DM_SOC_NSOC_diff.dat , DM.dat ;运行信息文件 out.fdf 。
程序标准输出: 因 \(NiO_{6}\) 输出矩阵过大,此处以相同配位环境下的单个Ni原子TB模型为例,介绍标准输出的部分内容,如下所示。
#=========================================#
Orbitals #第一列为轨道编号,第二列为原子编号,第三列为该原子内轨道编号
1 1 1
2 1 2
3 1 3
4 1 4
5 1 5
Total valence electrons number is: 8.00000
L,Theta,Phi: 2 0.000000000000 0.000000000000 #自旋矢量:球坐标形式
SOC Matrix: #将输入文件中的参数代入所得SOC矩阵
z2 up, x2-y2 up, yz up, zx up, xy up, z2 dn, x2-y2 dn, yz dn, zx dn, xy dn
0.000 0.000, 0.000 0.000, 0.000 0.000, 0.000 0.000, 0.000 0.000, 0.000 0.000, 0.000 0.000, 0.000 0.866, -0.866 0.000, 0.000 0.000
........................................(省略若干行)
TB+U WF: #考虑Hubbard排斥作用后的波函数矩阵
z2 up, x2-y2 up, yz up, zx up, xy up, z2 dn, x2-y2 dn, yz dn, zx dn, xy dn
0.000000 0.000000 0.000000 0.707107 0.000000 0.000000 0.000000 0.000000 0.707107 0.000000
........................................(省略若干行)
TB+U+SO WF in terms of the TB+U WF:
#在上述基础上加入SOC的波函数
#第一列为矩阵元所在行
#第二列为矩阵元所在列
#第三列为该矩阵元实部大小
#第四列为虚部大小
1 1 ( -0.362542236996 0.362542236996)
........................................(省略若干行)
H in the NSOC WF basis:
#不考虑SOC波函数基下的哈密顿量矩阵,每个括号内前一部分表示实部,后一部分表示虚部。
( -4.750 0.000) 0 ( -0.025 0.025) ( 0.043 -0.043) ( -0.000 -0.035) ( 0.035 -0.035) 0 ( 0.025 0.025) ( -0.035 -0.000) ( 0.043 0.043)
........................................(省略若干行)
Eigenvalues: #上述矩阵的本征值
1 -4.806103 0.000000
........................................(省略若干行)
Wavefunctions: #波函数数值
Basis: 1, 2, 3, 4, 5, 6, 7, 8, 9, 10,
WF 1 -0.03 -0.03, -0.01 0.01, -0.26 0.26, -0.26 0.26, 0.34 0.34, 0.04 -0.00, -0.00 0.02, 0.00 -0.36, -0.00 -0.36, -0.48 0.00,
........................................(省略若干行)
Diagonal Part of Density matrix: #密度矩阵对角元
Occ_Orb in Atom 1 1.007532836291 1.003016275637 1.994961542172 1.994961542172 1.999527803728 #1号原子各轨道占据数
Spin up #1号原子各轨道自旋向上占据数
0.503766418146 0.501508137819 0.997480771086 0.997480771086 0.999763901864
Spin down #1号原子各轨道自旋向下占据数
0.503766418146 0.501508137819 0.997480771086 0.997480771086 0.999763901864
........................................(省略若干行)
Total energy is: -25.514138174039 #体系的能量
#=========================================#
备注
体系的能量是将总的哈密顿量矩阵求解本征值后,按照从小到大顺序对本征值进行选取求和,选取本征值的数目等于轨道上实际的电子数,例如对于Ni,考虑SOC后3d轨道共有十条,对应哈密顿量矩阵为10*10矩阵,有10个本征值,实际上3d轨道有8个电子,则总能量为由小到大排序的前八个本征值求和所得。
2.4.3 手动加入哈密顿量的Slater-Koster紧束缚模型¶
PASP的S-K TB方法支持在原来总哈密顿量的基础上,添加额外的数值哈密顿量矩阵,需要在原 PASP.input 基础上加入“ TB.additional_H T ”的标签设置。并在原来两个输入文件之外,增加一个描述所要添加矩阵的文件“ Hmat_add.dat ”。以正八面体六配位环境下的单Ni原子示例,考虑SOC后,3d共有10个轨道,因此添加的哈密顿量矩阵也是10*10矩阵(注意矩阵元为复数)。输入文件实例在 “example_public/TB/SKTB_with_Hmat_add/input” 文件夹下。其中 Hmat_add.dat 文件内容如下:
#=========================================#
(0,0) (0,0) (0,0) (0,0) (0,0) (0,0) (0,0) (0,1.732) (-1.732,0) (0,0)
(0,0) (0,0) (0,0) (0,0) (0,2) (0,0) (0,0) (0,1) (1,0) (0,0)
(0,0) (0,0) (0,0) (0,-0.5) (0,0) (0,-1.732) (0,-1) (0,0) (0,0) (-0.5,0)
(0,0) (0,0) (0,0.5) (0,0) (0,0) (1.732,0) (-1,0) (0,0) (0,0) (0,0.5)
(0,0) (0,-2) (0,0) (0,0) (0,0) (0,0) (0,0) (0.5,0) (0,-0.5) (0,0)
(0,0) (0,0) (0,1.732) (1.732,0) (0,0) (0,0) (0,0) (0,0) (0,0) (0,0)
(0,0) (0,0) (0,1) (-1,0) (0,0) (0,0) (0,0) (0,0) (0,0) (0,-2)
(0,-1.732) (0,-1) (0,0) (0,0) (0.5,0) (0,0) (0,0) (0,0) (0,0.5) (0,0)
(-1.732,0) (1,0) (0,0) (0,0) (0,0.5) (0,0) (0,0) (0,-0.5) (0,0) (0,0)
(0,0) (0,0) (-0.5,0) (0,-0.5) (0,0) (0,0) (0,2) (0,0) (0,0) (0,0)
#=========================================#
备注
添加矩阵的大小应和PASP生成矩阵大小相同;
添加矩阵时,各轨道对应的矩阵元要注意按照PASP中TB的轨道顺序进行添加(关于轨道顺序,参见2.4.2节“哈密顿量矩阵输出文件”部分的相关内容);
根据Fortran读取复数的语法规则,括号内实部与虚部之间需要逗号隔开;
添加的矩阵应是厄密的,PASP会对矩阵的厄密性进行检查。
2.5 自旋序诱导铁电性统一极化模型¶
2.5.1 自旋序诱导铁电极化的理论模型简介¶
1994年瑞士的Schmid明确提出了多铁性材料(multi-ferroic)的概念,即具有两种或两种以上初级铁性体特征的单相化合物。磁致多铁材料是多铁性材料大家族中的后起之秀,其特色在于其铁电性起源于特定的磁序,因此其铁电与磁性紧密关联,具有本征的强磁电耦合效应。Khomskii根据物理机制对多铁性材料进行分类,他将上述磁致多铁材料单独列为一列,称为第二类(type-II)多铁体,其余体系统统归为一类,称为第一类(type-I)多铁体。
从对称性的角度看,铁电性破坏空间反演对称性。如果晶体结构本身满足空间反演对称,但其磁结构破坏了空间反演对称,则该磁序则有可能导致铁电极化。2005年以来,Nagaosa、Dagotto等人提出一些微观模型部分解释了磁序诱导铁电性现象,但是这些模型还存在一些问题。2011年以来,向红军等人提出了一个用来描述磁序诱导铁电性的统一极化模型。该模型认为自旋序诱导的铁电极化包含两部分: \(\vec{P}_{t} \approx \vec{P}_{e}\left(\vec{S}_1, \vec{S}_2, \ldots,\vec{S}_m;\vec{U}=0;\Theta=0\right)+\vec{P}_{ion,lattice}(\vec{U};\Theta)\) ,这里 \(\vec{S}_{i}\) 表示磁性离子的自旋, \(\vec{U}=\left(\vec{u}_1,\vec{u}_3,\cdots,\vec{u}_n \right)\) 表示离子相对参考结构(一般是高温中心对称结构)的位移, \(\Theta=\left(\eta_1,\eta_2, \cdots,X_6\right)\) 表示相对参考结构的应变。其中纯电子部分 \(\vec{P}_{e}\) 描述自旋序引起的电荷重新分布而诱导的铁电极化,离子-晶格部分 \(\vec{P}_{\text {ion,lattice}}\) 描述自旋-晶格耦合导致的离子位移和晶格形变而导致的铁电极化。对于自旋序诱导的铁电极化的纯电子部分,一个磁性离子对诱导的电子极化是这两个磁性离子自旋的二次函数: \(\vec{P}_{e}={\textstyle \sum_{i=1}^{2}}{\textstyle\sum_{\alpha \beta}}\vec{P}_{i}^{\alpha \beta}S_{i \alpha}S_{i \beta}+{\textstyle \sum_{\alpha \beta}}\vec{P}_{12}^{\alpha \beta}S_{1 \alpha}S_{2 \beta}\) ,其中第一项是由极化参数矩阵 \(\vec{P}_{i}^{\alpha \beta}\) 决定的与单个自旋有关的单格点项(intra-site),而第二项是由极化参数矩阵 \(\vec{P}_{12}^{\alpha \beta}\) 决定的与两个自旋都有关的格点间项(inter-site)。
为了得到极化的离子-晶格部分贡献,需要计算由于自旋序导致极性离子位移和晶格形变。一般地,自旋-晶格耦合体系的总哈密顿包含两部分: \(\mathrm{E}\left(u_{m}, \eta_{j}, \vec{S}_{i}\right)=E_{P M}\left(u_{m}, \eta_{j}\right)+E_{s p i n}\left(u_{m}, \eta_{j}, \vec{S}_{i}\right)\) 。 \(E_{P M}\left(u_{m}\right)\) 是与自旋无关的顺磁态的弹性能部分, \(E_{PM}=E_{0}+{\textstyle \sum_{m}}A_{m}u_{m}+{\textstyle \sum_{j}}A_{j}\eta_{j}+\frac{1}{2}{\textstyle \sum_{m,n}}B_{mn}u_{m}u_{n}+\frac{1}{2}{\textstyle \sum_{j,k}} B_{jk}\eta_{j}\eta_{k}\) \(+{\textstyle \sum_{m,j}}B_{mj}u_{m}\eta_{j}+O(3)\) . \(E_{0}\) 是参考态 \(\left(u_{m}=0, \eta_{j}=0\right)\) 能量。通常假定参考态处于平衡态(原子受力和应力为0), \(A_{m}=0, A_{j}=0\) 。
\(E_{spin}\) 是自旋相互作用哈密顿,包含对称交换相互作用 \(E_{H}\) ,反对称DM相互作用 \(E_{DM}\) ,和单离子各向异性项: \(E_{S I A}:E_{\text {spin}}=E_{H}+E_{D M}+E_{S I A}\) 。其中对称交换相互作用 \(E_{H}={\textstyle \sum_{i,i}}J_{i,i'}^{0}\vec{S}_{i}\cdot \vec{S}_{i'}+{\textstyle \sum_{i,i'}}\frac{\partial J_{i,i'} }{\partial u_{m}}\vec{S}_{i}\cdot \vec{S}_{i'}u_{m}+{\textstyle \sum_{i,i'}}\frac{\partial J_{i,i'}}{\partial \eta_{j}}\vec{S}_{i}\cdot \vec{S}_{i}\eta_{j}+O(3)\) ( \(E_{DM}\) 和 \(E_{SIA}\) 的表达式与 \(E_{H}\) 类似), \(J_{i,i'}^{0}\) , \(\frac{\partial J_{i,i'} }{\partial u_{m}}\) , \(\frac{\partial J_{i,i'}}{\partial \eta_{j}}\) 都可以利用四态法计算。通过求总能量极小值,可以计算出特定磁序下的离子的极性位移: \(\left\{\begin{array}{l}\frac{\partial \mathrm{E}\left(u_{m}, \eta_{j}, \vec{S}_{i}\right)}{\partial u_{m}}=0 \\\frac{\partial \mathrm{E}\left(u_{m}, \eta_{j}, \vec{S}_{i}\right)}{\partial \eta_{j}}=0\end{array}\right.\) 。然后利用玻恩有效电荷 \(Z_{\alpha m}\) 和压电系数 \(e_{\alpha j}\) ,可以得到极化的离子位移贡献: \(\mathrm{P}_{\alpha}={\textstyle \sum_{m}}\mathrm{Z}_{\alpha \mathrm{m}}u_{m}+\sum_{j}\mathrm{e}_{\alpha \mathrm{j}}\eta_{j}\) 。力常数 \(B_{mn}\) ,弹性常数 \(B_{jk}\) ,internal displacement tensor \(B_{mj}\) , \(z_{\alpha m}\) , \(e_{\alpha j}\) 可以利用密度泛函微扰理论或有效差分方法计算得到。
2.5.2 自旋序诱导铁电极化的计算流程¶
2.5.2.1 铁电极化的纯电子贡献¶
以下是计算铁电极化的纯电子贡献时, PASP.input 部分关键设置项的例子。完整示例输入文件见 “example_public/P_by_spin_order/pure_electronic/input” 文件夹。
#=========================================#
Model KNB
%block isaAB
2 2
%endblock isaAB
fig.maxk 2 #考虑自旋离子对
%block fig.mdist
1 11
%endblock fig.mdist
GKNB.symP T #是否在程序里根据晶体结构对称性来对称化极化参数
%block GKNB_inter_site
1 #不等价离子对的个数
6.184071509801 6.184071509801 6.184071509801 #Fe2
4.638053632351 6.184071509801 9.276107264702 #Fe35
1 0 0 #局域坐标系x轴
0 1 0 #局域坐标系y轴
0 0 1 #局域坐标系z轴
0.001254 0.00213 0.002374 #xx
0 0 0 #xy
0 0 0 #xz
0 0 0 #yx
0.001254 0.00213 0.002374 #yy
0 0 0 #yz
0 0 0 #zx
0 0 0 #zy
0.001254 0.00213 0.002374 #zz
%endblock GKNB_inter_site
%block GKNB_single_site
1
6.184071509801 6.184071509801 6.184071509801 #Fe2
1 0 0 #局域坐标系x轴
0 1 0 #局域坐标系y轴
0 0 1 #局域坐标系z轴
0 #Pxx
0 #Pyy
0 #Pzz
0 #Pxy=Pyx
0 #Pxz=Pzx
0 #Pyz=Pzy
%endblock GKNB_single_site
#=========================================#
其中,“Model KNB”表示采用了KNB模块。“ block isaAB ”设定磁性离子的元素种类,目前最多可以考虑两种磁性离子,这个例子表示第二种元素是磁性离子。“ block fig.mdist ”里第二个数(这里的11)表示考虑磁性离子对之间的最大距离(以Bohr为单位)。“ block GKNB_inter_site ”设定格点间项极化参数矩阵 \(\vec{P}_{12}^{\alpha\beta}\) 。这个block里第一行给出了不等价离子对的个数。接下来需要对每一个不等价离子对给出离子位置的直角坐标(Å为单位),采用的局域坐标系(通常为单位矩阵),和 \(\vec{P}_{12}^{\alpha\beta}\) 分量。单格点项极化参数矩阵 \(\vec{P}_{12}^{\alpha\beta}\) 的输入方式与格点间项极化参数矩阵类似(对于目前例子为0,可以不设定)。另外,体系的磁序由 spin3.in 文件设定。
运行PASP以后,标准输出会给出极化大小,比如:
spin_FE: P is 0.000000019290 0.000000000000 0.000000000000 muC/m^2
2.5.2.2 极化的离子-晶格部分贡献¶
以下是计算铁电极化的离子-晶格部分贡献时, PASP.input 部分关键设置项的例子。完整示例输入文件见 “example_public/P_by_spin_order/ion_strain/input” 文件夹。
#=========================================#
Model Spin_lattice_interaction
spin_lat.spin_force T #计算自旋序导致的离子受力
spin_lat.spin_int_stress T #计算自旋序导致的晶格应力
Read_init_spin T #从spin3.in文件读入磁序
phonopy_force_constant T #读入phonopy给出的力常数文件
phon.sym_born_chg T #是否对称化给定的波恩有效电荷张量
#=========================================#
除了需要准备通常的输入文件以外,还需要给出 spin_exchange.dat (包含磁相互作用及海森堡磁相互作用对位移的偏导 \(\frac{\partial J_{i,i'}}{\partial u_{m}}\) ,格式规则见3.3节), SPOSCAR (phonopy里计算力常数用的超胞), FORCE_CONSTANTS (phonopy给出的力常数), Zeff.dat (波恩有效电荷张量,见3.12节), elastic.dat (包含弹性常数, internal displacement tensor ,压电张量,见3.13节), spin_strain.dat (包含海森堡磁相互作用对应变的偏导 \(\frac{\partial J_{i,i{}'}}{\partial \eta _{j}} \) 文件设定DM相互作用对位移的偏导(见3.15节)。
运行PASP以后,标准输出会给出极化大小,比如:
Total P_{ion+lattice} (muC/m^2): -4.578990691511 2.943211932078 -11595.182705833875
2.6 线性自旋波色散的计算¶
2.6.1 线性自旋波理论简介¶
自旋波(spin wave)是由于电子自旋之间存在的交换作用,在磁体中自发产生的一种元激发,又称为磁振子(magnon)。1936年,布洛赫(F.Bloch)提出自旋波理论,用来解释低温下磁化强度随着温度变化的关系。在20世纪50年代,通过磁共振和中子衍射方法,从实验中证实了自旋波的存在。目前PASP可以计算一般性二次自旋哈密顿量的自旋波色散:
其中 \(m,n\) 是晶胞序号, \(i,j\) 代表晶胞中的磁性离子编号, \(J\) 和 \(A\) 分别代表自旋相互作用张量和单离子各向异性张量(都用矩阵表示)。利用Holstein–Primakoff变换可以把自旋算符表示成玻色算符,保留玻色算符最高到二阶项,对玻色算符作傅里叶变换,可以得到不同 \(q\) 点二阶玻色子哈密顿矩阵 \(h_{q}\) (具体可参考文献 16 )。利用Colpa提出的方法 17 ,可以对角化该二阶玻色子哈密顿矩阵,从而得到自旋波色散。
2.6.2 线性自旋波色散的计算流程¶
以下是计算线性自旋波色散时, PASP.input 部分关键设置项的例子。完整示例输入文件见 “example_public/magnon/input” 文件夹。
#=========================================#
Model magnon
Read_init_spin T
spin_lat.general_J T
number_of_qpoints 100
%block qpoints.magnon
0.5 0.000000000000 0.
0.0000 0.0000000 0.
%endblock qpoints.magnon
#=========================================#
其中,“Model magnon”代表采用计算线性自旋波色散的模块。“Read_init_spin T”表示基态磁序(可以是非共线磁基态)从 spin3.in 文件中读取。自旋相互作用参数从 spin_exchange.dat 文件读取(参见3.3节格式说明,注意须采用矩阵形式。另外注意该模块暂未支持四阶点乘相互作用,请勿添加这类相互作用信息)。“spin_lat.general_J T”表示自旋相互作用采用了一般性的二次相互作用形式。目前PASP每次计算布里渊区1条线段上的自旋波色散,其中“number_of_qpoints”代表这条线段上q点的数量,“block qpoints.magnon”里第一行代表这条线段的起始q点(q用以倒格矢展开的系数 \(q_{i}\) 表示: \(q=\textstyle \sum_{i}q_{i}b_{i}\) ),第二行代表这条线段的终末q点。
运行PASP以后,输出文件 spin_wave_E.dat 会提供这条线段上q点对应的自旋波能量,该文件可以用xmgrace等软件可视化。
- 1
LOU F, LI X Y, JI J Y, et al. Pasp: Property analysis and simulation package for materials [J]. Journal of Chemical Physics, 2021, 154(11):
- 2
The vasp manual - vaspwiki [M]. https://www.vasp.at/wiki/index.php/The_VASP_Manual.
- 3
LI X-Y, LOU F, GONG X-G, et al. Constructing realistic effective spin hamiltonians with machine learning approaches [J]. New Journal of Physics, 2020, 22(5):
- 4
XIANG H, WEI S-H, GONG X. Structures of [ag7 (sr) 4]− and [ag7 (dmsa) 4]− [J]. Journal of the American Chemical Society, 2010, 132(21): 7355-7360.
- 5
LU X-Z, RONDINELLI J M. Epitaxial-strain-induced polar-to-nonpolar transitions in layered oxides [J]. Nature materials, 2016, 15(9): 951-955.
- 6
WANG H, TANG F, STENGEL M, et al. Convert widespread paraelectric perovskite to ferroelectrics [J]. Physical review letters, 2022, 128(19): 197601.
- 7
BAYARAA T, XU C, BELLAICHE L. Magnetization compensation temperature and frustration-induced topological defects in ferrimagnetic antiperovskite mn 4 n [J]. Physical Review Letters, 2021, 127(21): 217204.
- 8
LI X, WU X, YANG J. Room-temperature half-metallicity in la (mn, zn) aso alloy via element substitutions [J]. Journal of the American Chemical Society, 2014, 136(15): 5664-5669.
- 9
XIANG H J, KAN E J, WEI S-H, et al. Predicting the spin-lattice order of frustrated systems from first principles [J]. Physical Review B, 2011, 84(22):
- 10
XIANG H, LEE C, KOO H-J, et al. Magnetic properties and energy-mapping analysis [J]. Dalton Transactions, 2013, 42(4): 823-853.
- 11(1,2)
HUKUSHIMA K, NEMOTO K. Exchange monte carlo method and application to spin glass simulations [J]. Journal of the Physical Society of Japan, 1996, 65(6): 1604-1608.
- 12(1,2)
EARL D J, DEEM M W. Parallel tempering: Theory, applications, and new perspectives [J]. Physical Chemistry Chemical Physics, 2005, 7(23): 3910-3916.
- 13(1,2)
HANSMANN U H E. Parallel tempering algorithm for conformational studies of biological molecules [J]. Chemical Physics Letters, 1997, 281(1-3): 140-150.
- 14
WHITNEY A W. Direct method of nonparametric measurement selection [J]. Ieee Transactions on Computers, 1971, C 20(9): 1100-1103.
- 15(1,2)
MIYATAKE Y, YAMAMOTO M, KIM J J, et al. On the implementation of the heat bath algorithms for monte-carlo simulations of classical heisenberg spin systems [J]. Journal of Physics C-Solid State Physics, 1986, 19(14): 2539-2546.
- 16
TOTH S, LAKE B. Linear spin wave theory for single-q incommensurate magnetic structures [J]. Journal of Physics: Condensed Matter, 2015, 27(16): 166002.
- 17
COLPA J. Diagonalization of the quadratic boson hamiltonian [J]. Physica A: Statistical Mechanics and its Applications, 1978, 93(3-4): 327-353.
- 18
MIYATAKE Y, YAMAMOTO M, KIM J J, et al. On the implementation of the heat bath algorithms for monte-carlo simulations of classical heisenberg spin systems [J]. Journal of Physics C-Solid State Physics, 1986, 19(14): 2539-2546.
- 19
NI J, LI X, AMOROSO D, et al. Giant biquadratic exchange in 2d magnets and its role in stabilizing ferromagnetism of nicl 2 monolayers [J]. Physical Review Letters, 2021, 127(24): 247204.