4. 应用实例¶
4.1. Nanodcal 实例¶
以Benzene分子的自洽、本征态计算为例详细描述 Nanodcal 在Device Studio中的应用。包含创建项目,从建模到计算,到数据的可视化分析整个流程。实际上, Nanodcal 的功能不仅于此,详情可参照 Nanodcal 应用教程或点击对应的紫色字体软件名称。Device Studio可以生成 Nanodcal 很多功能计算的输入文件,用户可根据计算需要选择生成。
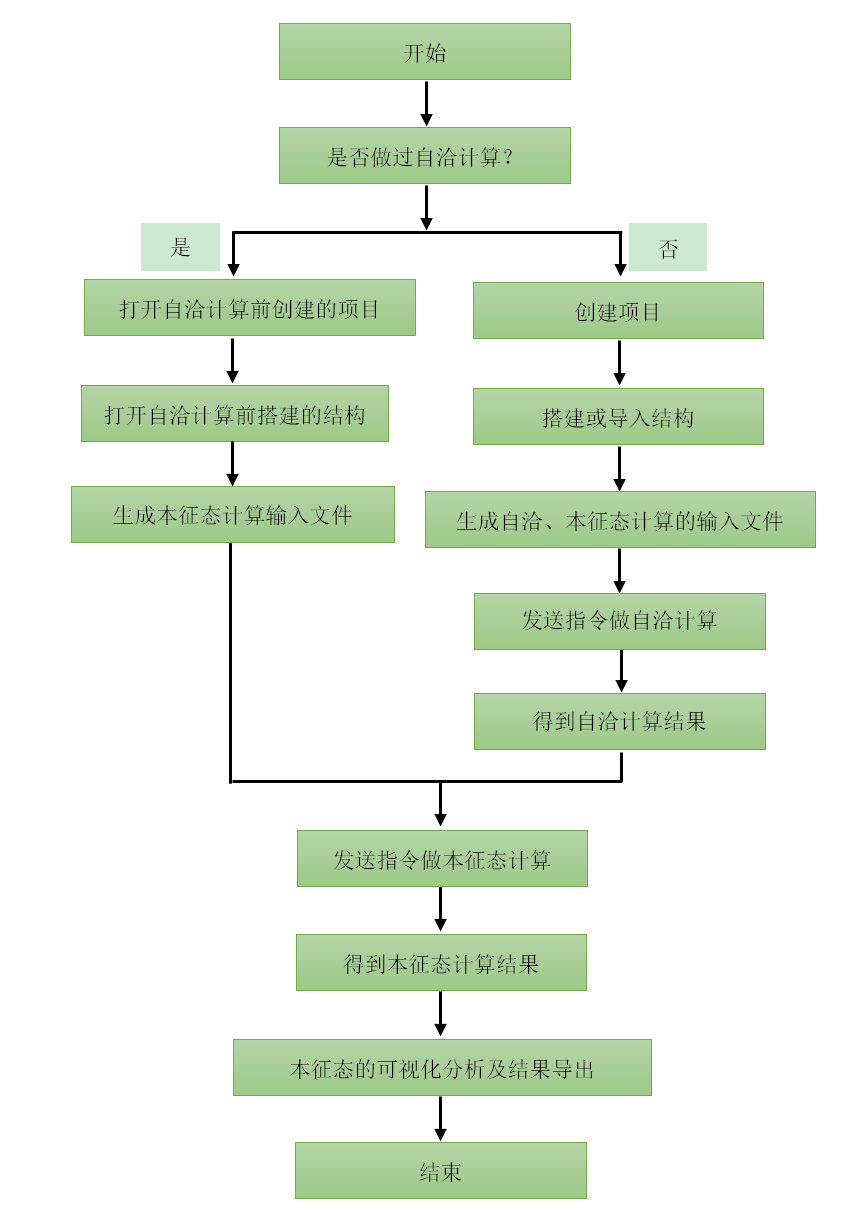
4.1.1. 计算流程¶

4.1.2. 创建项目¶
双击Device Studio图标快捷方式启动软件,其图形界面如图3-1所示,根据界面提示选择创建一个新的项目(Create a new Project)或打开一个已经存在的项目(Open an existing Project)的按钮,选中之后点击界面中的 OK 按钮即可。若选择创建一个新的项目,用户可根据需要给该项目命名,如本项目命名为 Nanodcal,或采用软件默认项目名。
4.1.3. 搭建或导入结构¶
对于想要搭建的结构,用户可先在Device Studio本地数据库或在线数据库搜索结构是否存在。若存在,找到结构直接导入;若不存在,可将库中的相关结构先导入,在此基础上方便快捷的搭建,也可直接自行搭建。如Benzene分子,该结构在Device Studio本地库中存在,则可在本地库中找到该结构直接导入即可,导入Benzene分子结构后的图形界面如图4-1-3所示。在Device Studio中导入Benzene分子结构操作这里不做详细说明,用户可参照 导入结构 节内容。
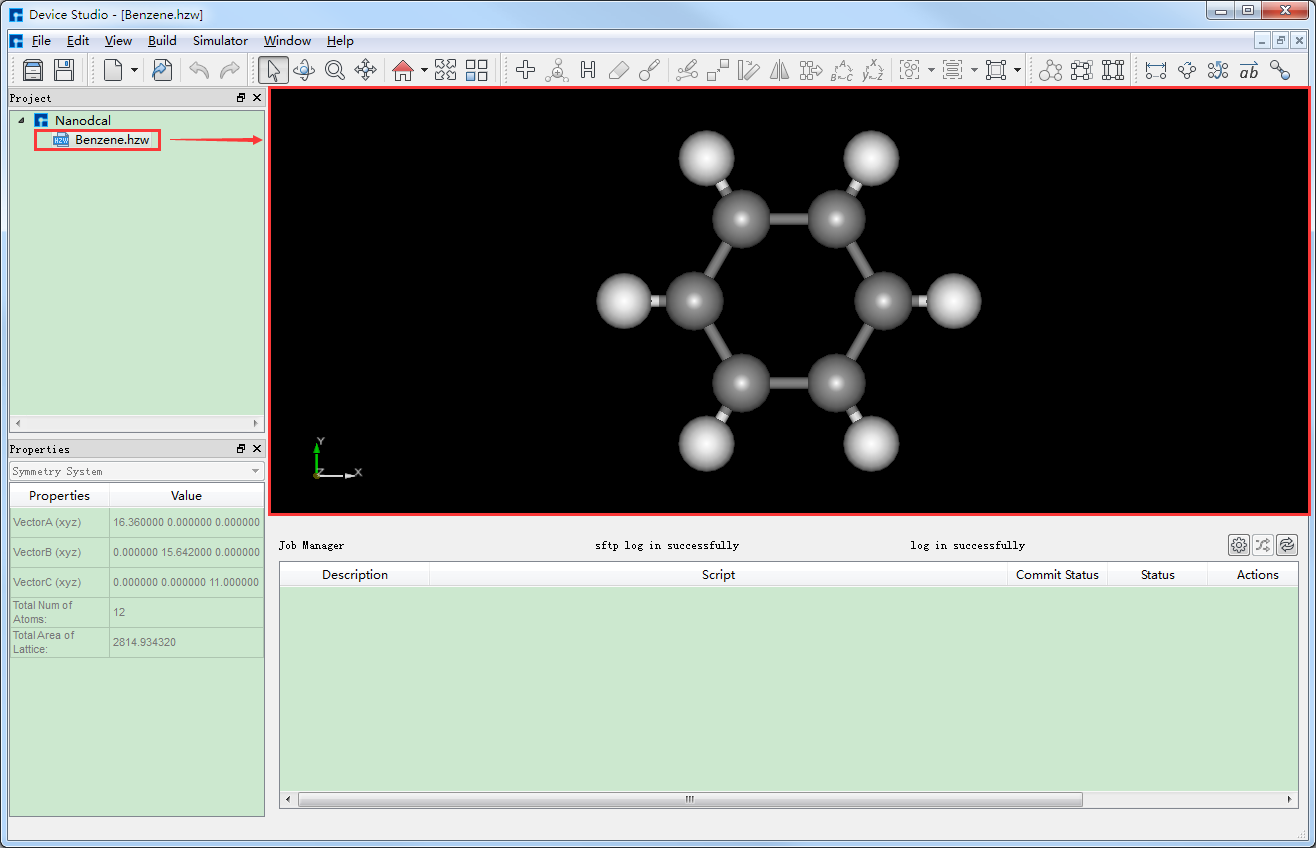
图 4-1-3: 导入Benzene分子结构后的图形界面¶
4.1.4. 生成自洽计算输入文件¶
在如图4-1-3所示界面中选中 Simulator → Nanodcal → SCF Calculation,弹出界面如图4-1-4(a)所示,用户可根据所计算的结构及计算需要分别选中 Basic settings 、Iteration control、Basis set、Spin type 这四个按钮合理设置参数,之后点击 Generate files 即可生成自洽计算输入文件。
如生成Benzene分子自洽计算输入文件,根据计算需要设置参数,分别选中 Basic settings 、Iteration control、Basis set、Spin type 设置参数分别如图4-1-4(a)、4-1-4(b)、4-1-4(c)和4-1-4(d)所示,设置好参数后点击 Generate files 即可生成Benzene自洽计算输入文件 scf.input。
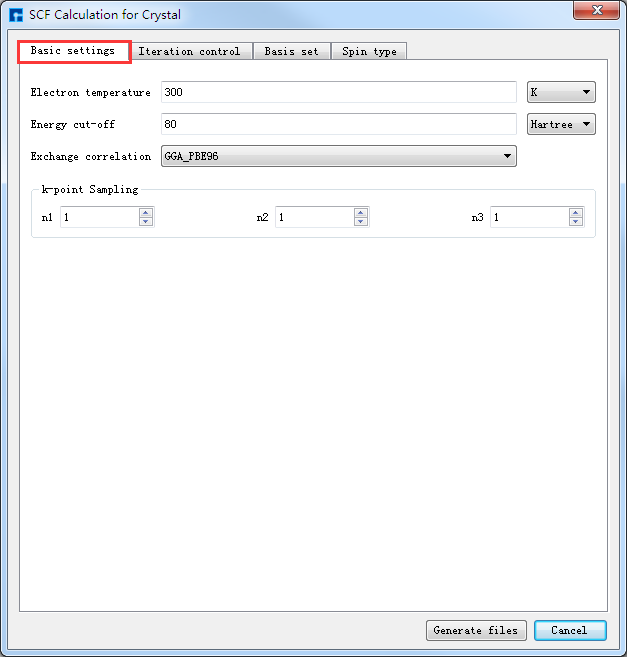
图 4-1-4(a)¶ |
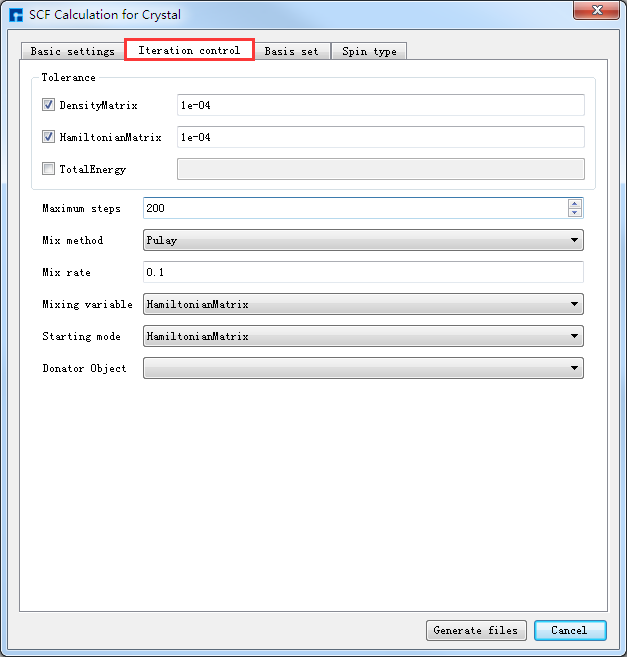
图 4-1-4(b)¶ |
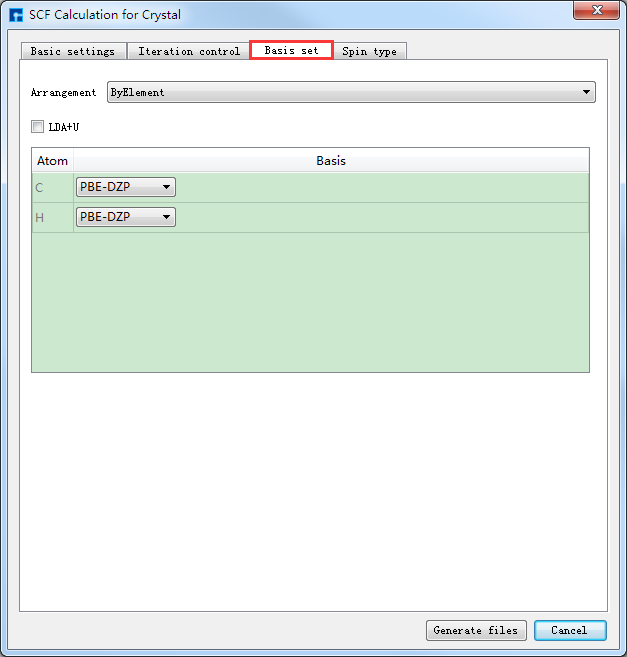
图 4-1-4(c)¶ |
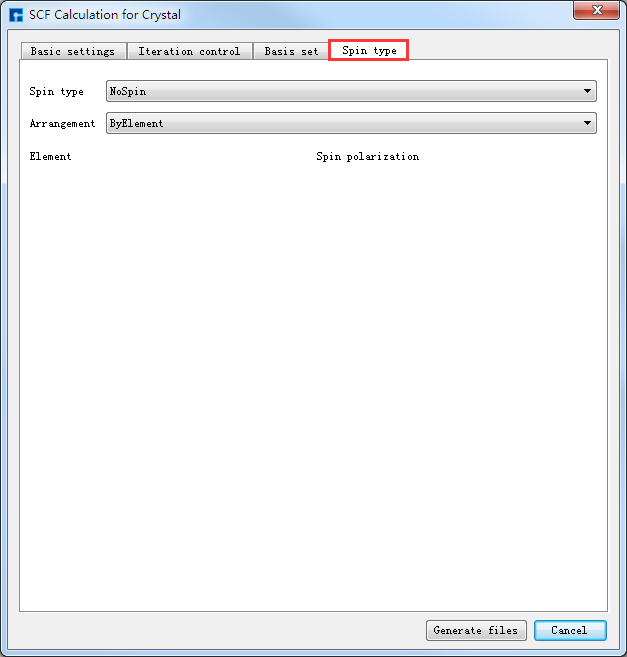
图 4-1-4(d)¶ |
4.1.5. 生成本征态计算输入文件¶
在如图4-1-3所示界面中选中 Simulator → Nanodcal → Analysis,弹出Analysis界面,在界面中双击 EigenStates,并设置参数如图4-1-5所示,设置好参数后点击 Generate files 即可生成Benzene本征态计算输入文件 EigenStates.input。
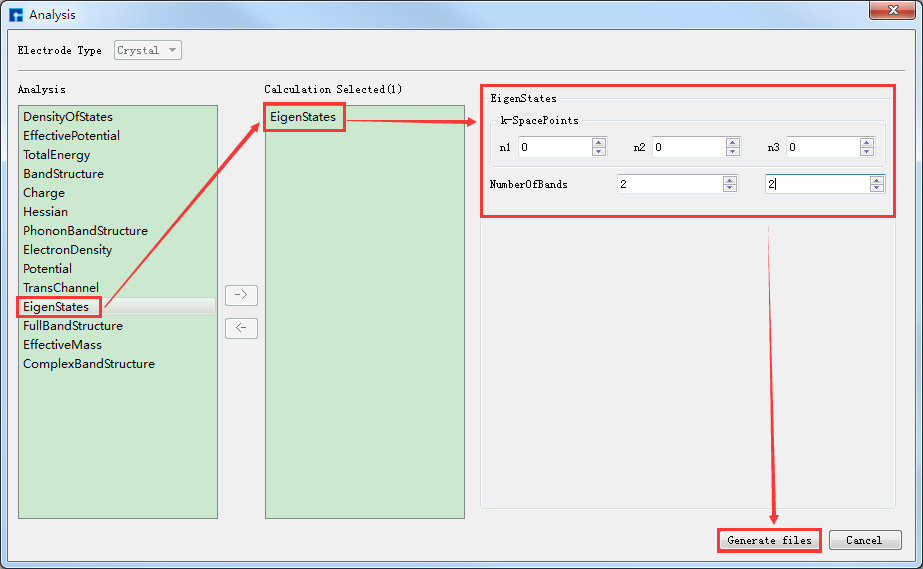
图 4-1-5: 设置Benzene分子本征态计算参数界面¶
4.1.6. 连接服务器¶
生成输入文件之后需要计算,计算之前需连接装有Nanodcal的服务器。在如图4-1-6所示界面中点击 设置 按钮,弹出MachineOptions界面,在该界面中点击 New 按钮,弹出MachineSet界面,在该界面中填写Computer Name、HostIp、part、Username、Password等一系列信息,点击 OK 按钮则在MachineOptions界面中添加了装有Nanodcal的服务器如图4-1-7所示。在图4-1-7所示的界面中选中装有Nanodcal的服务器,点击 Select 按钮则连接上装有Nanodcal的服务器。同理,若要连接装有Nanodcal本地电脑,在图4-1-7所示界面中选中MyComputer,点击 Select 即可。
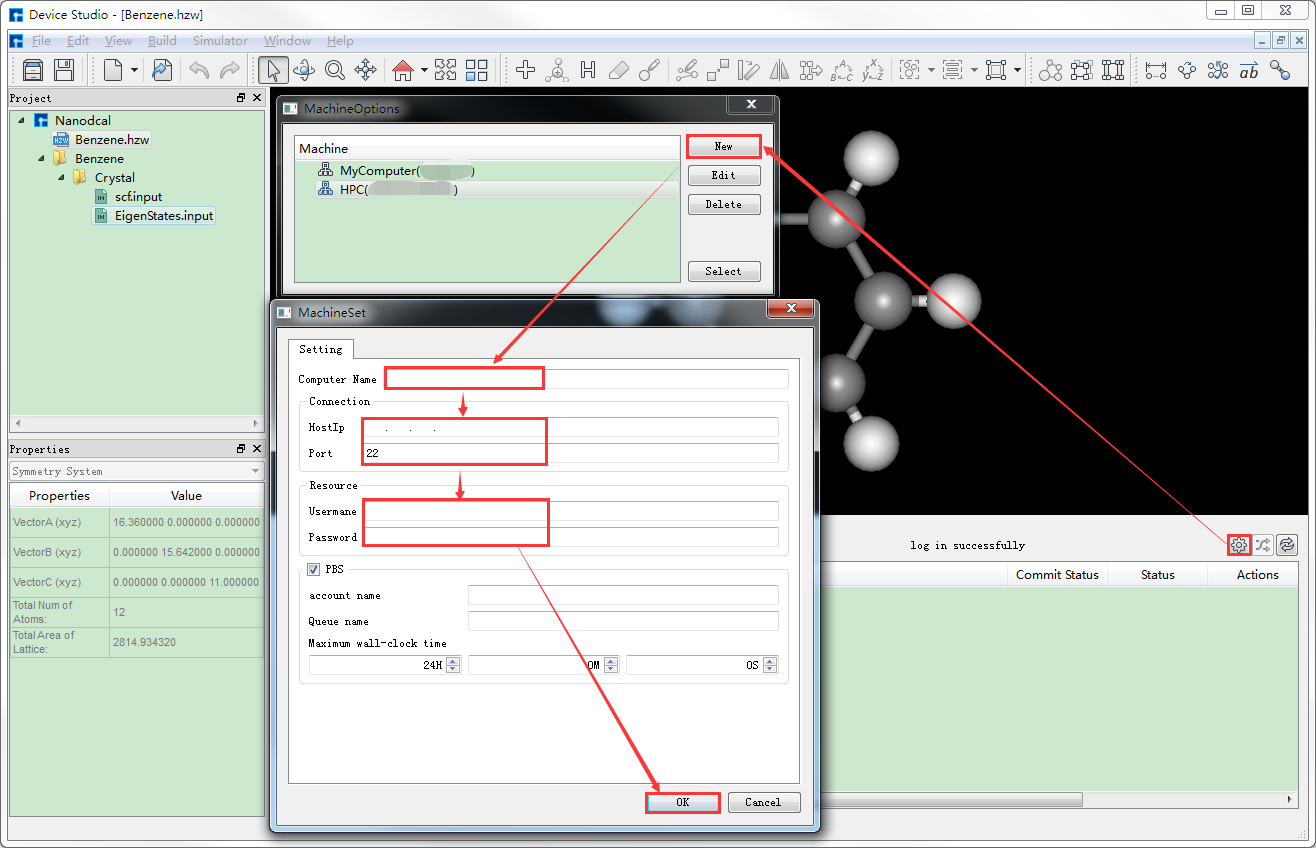
图 4-1-6: 连接服务器操作界面¶
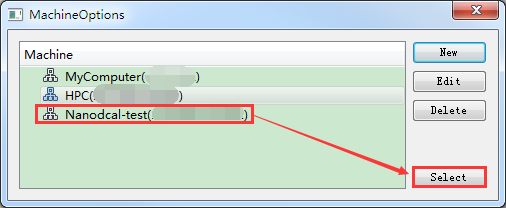
图 4-1-7 MachineOptions界面¶
4.1.7. 自洽计算¶
在做自洽计算之前需查看计算所需的输入文件(scf.input)和基组文件(C_PBE-DZP.nad 和 H_PBE-DZP.nad)是否齐全,再根据计算需要打开 scf.input 文件并查看文件中的参数设置是否合理,若不合理,则可选择直接在文件中进行编辑或重新生成,最后再进行自洽计算。具体步骤如下:
(1)连接装有Nanodcal的服务器。
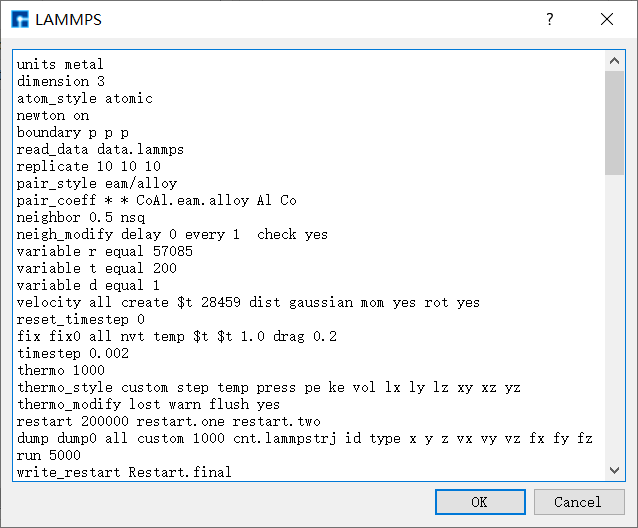
(2)检查自洽计算所需的输入文件(scf.input)和基组文件(C_PBE-DZP.nad 和 H_PBE-DZP.nad)是否齐全。选中 scf.input → 右击 → Open with 即可查看到Benzene分子自洽计算输入文件如下所示:
%%What quantities should be calculated
calculation.name = scf
%Basic setting
calculation.occupationFunction.temperature = 300
calculation.realspacegrids.E_cutoff = 80 Hartree
calculation.xcFunctional.Type = GGA_PBE96
calculation.k_spacegrids.number = [ 1 1 1 ]'
system.centralCellVectors = [[16.36 0 0]' [0 15.642 0]' [0 0 11]']
system.spinType = NoSpin
%Iteration control
calculation.SCF.monitoredVariableName = {'rhoMatrix','hMatrix','totalEnergy','bandEnergy','gridCharge','orbitalCharge'}
calculation.SCF.convergenceCriteria = {1e-04,1e-04,[],[],[],[]}
calculation.SCF.maximumSteps = 200
calculation.SCF.mixMethod = Pulay
calculation.SCF.mixRate = 0.1
calculation.SCF.mixingMode = H
calculation.SCF.startingMode = H
%calculation.SCF.donatorObject = NanodcalObject.mat
%Basic set
system.neutralAtomDataDirectory = '../'
system.atomBlock = 12
AtomType OrbitalType X Y Z
C PBE-DZP 5.00900000 5.57200000 5.00000000
C PBE-DZP 5.77900000 4.23800000 5.00000000
C PBE-DZP 7.31900000 4.23800000 5.00000000
C PBE-DZP 8.08900000 5.57200000 5.00000000
C PBE-DZP 7.31900000 6.90500000 5.00000000
C PBE-DZP 5.77900000 6.90500000 5.00000000
H PBE-DZP 3.86900000 5.57200000 5.00000000
H PBE-DZP 5.20900000 3.25100000 5.00000000
H PBE-DZP 7.88900000 3.25100000 5.00000000
H PBE-DZP 9.22900000 5.57100000 5.00000000
H PBE-DZP 7.88900000 7.89200000 5.00000000
H PBE-DZP 5.21000000 7.89300000 5.00000000
end
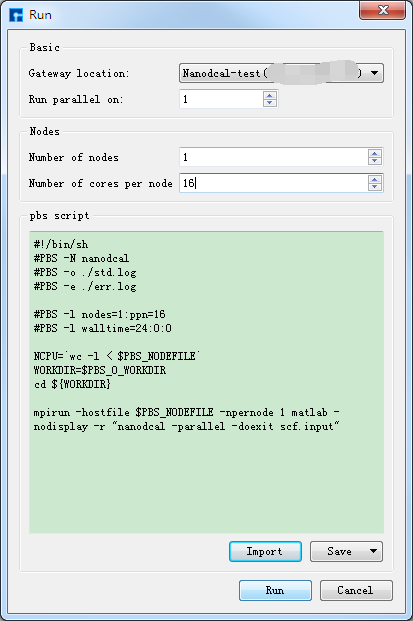
图 4-1-8 Run界面¶
(3)在Device Studio的Project Explorer区域选中 scf.input → 右击 → Run,弹出Run界面如图4-1-8所示。根据计算需要设置参数后点击 Save 按钮保存相应的Pbs脚本,之后点击如图4-1-8所示界面中的 Run 按钮则可做Benzene分子的自洽计算。用户可在Job Manager区域中观测自洽计算状态,当自洽计算任务处于排队中、计算中和计算完成时,Status 分别为Queued、Running、Finished;在计算过程中,点击 刷新 按钮,通过 Commit Status 、Status 可查看到自洽计算的计算进度和计算状态;计算完成后Device Studio的Job Manager区域如图4-1-9所示,点击 Action 下的 下载 按钮弹出下载Benzene分子自洽计算结果界面如图4-1-10所示,在该界面找到自洽计算的结果文件 NanodcalObject.mat,点击 Download 则可下载,下载后可在软件的Project Explorer区域查看到该结果文件。
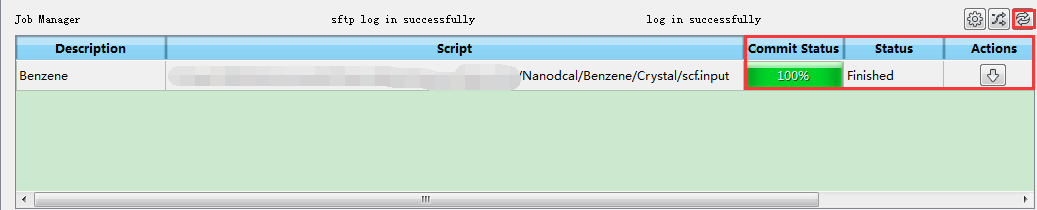
图 4-1-9 Benzene分子自洽计算完成的Device Studio的Job Manager区域¶
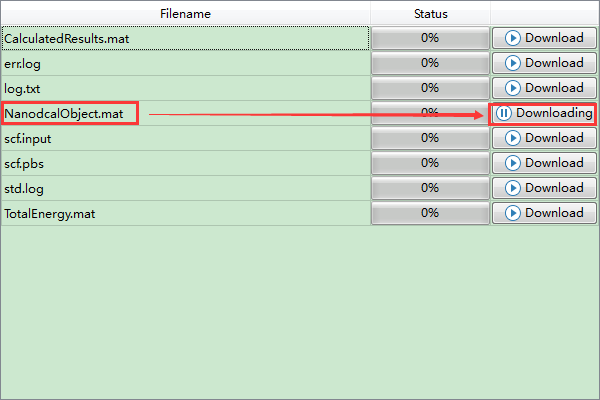
图 4-1-10 下载Benzene分子自洽计算结果界面¶
4.1.8. 本征态计算¶
本征态计算需调用自洽计算结果,故需先做自洽计算,再做本征态计算。本算例已经做过自洽计算,则不需要再做自洽计算,直接调用自洽计算结果做本征态计算即可。Benzene分子的本征态计算的具体步骤如下:
(1)连接装有Nanodcal的服务器。
(2)选中 EigenStates.input → 右击 → Open with 即可查看到Benzene分子本征态计算输入文件如下所示:
system.object = NanodcalObject.mat
calculation.name = eigenStates
calculation.eigenStates.kSpacePoints = [0 0 0]'
calculation.eigenStates.numberOfBands = [2,2]
calculation.eigenStates.realSpace = true
calculation.eigenStates.plot = true
%calculation.control.xml = true
calculation.control.dsf = 1
(3)在Device Studio的Project Explorer区域选中 EigenStates.input → 右击 → Run,弹出Run界面如图4-1-8所示。根据计算需要设置参数后点击 Save 按钮保存相应的Pbs脚本,点击界面中的 Run 按钮则可做Benzene分子的本征态计算,计算完成后下载本征态计算结果文件 EigenStates.dsf。
4.1.9. 本征态的可视化分析¶
对于本征态的可视化分析有3种情况,分别为3维(3D)、2维(2D)和1维(1D),3种可视化分析可自由切换,其中2D的可视化分析又分为平面显示和立体显示2种情况,用户可根据需要进行选择。
4.1.9.1. 本征态的3D可视化分析¶
在Device Studio的Project Explorer区域选中本征态计算结果文件 EigenStates.dsf → 右击 → Show View,弹出本征态的3D可视化分析界面如图4-1-11,由图4-1-11可知,用户可根据需要选择在可视化分析结果中是否显示该体系的结构信息以及显示该结构信息的方式;可选择是否显示Lattice框以及显示Lattice框的方式(分子结构没有Lattice框,故本算例中无法使用该功能);可通过改变Isovalue值来改变本征态3D可视化分析的形状;可通过选择如图4-1-11中所示的红色框选部分来选择从不同平面来观察本征态的3D可视化分析结果;可通过滚动鼠标中键将3D可视化分析结果放大或缩小;可按住鼠标右键,通过拖动鼠标将3D可视化分析结果进行旋转;可根据需要选择不同的Colormap。在如图4-1-11所示的界面中BandValues部分,用户可通过下拉选择显示第几条带的本征态可视化分析结果。
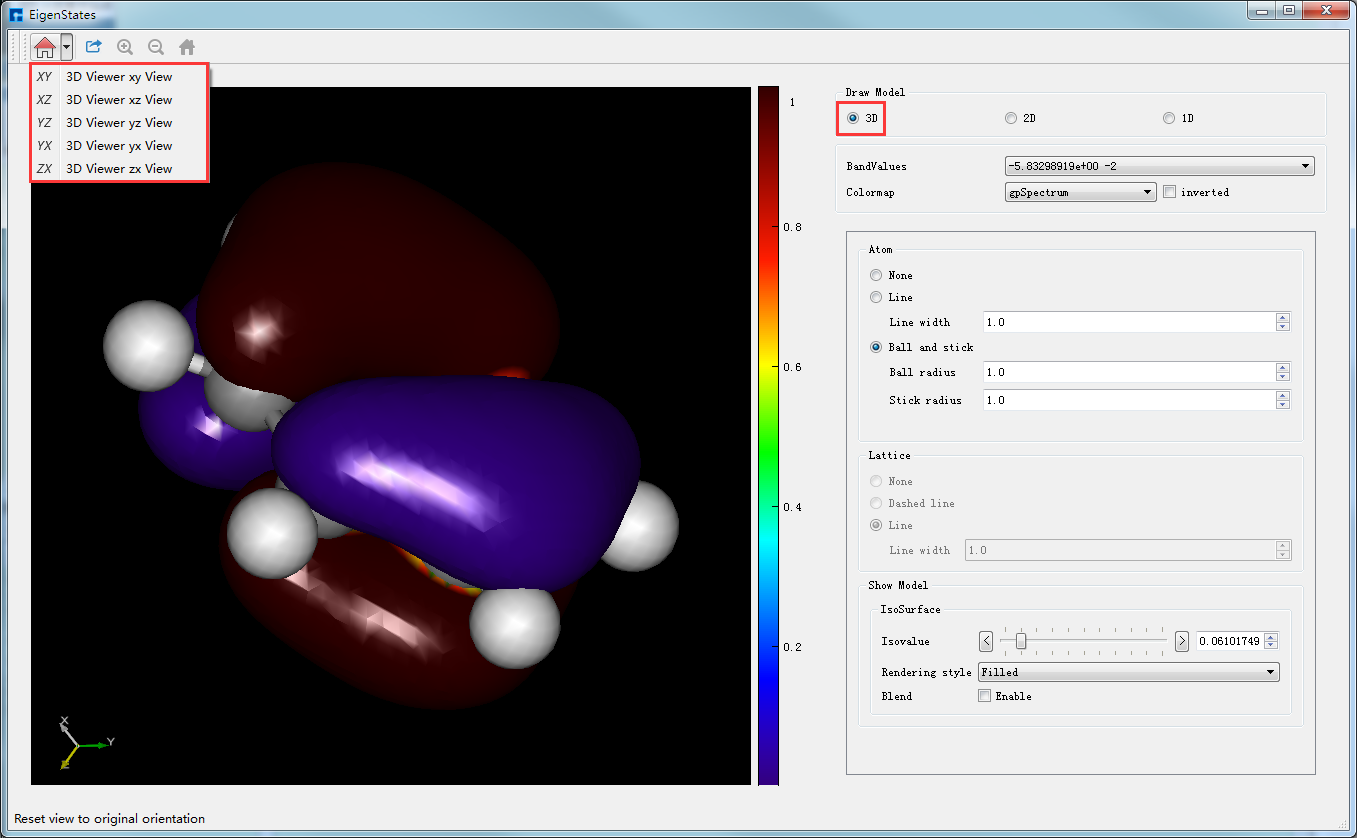
图 4-1-11 Benzene分子本征态的3D可视化分析界面¶
选中如图4-1-11所示界面中 Export 快捷图标,弹出导出可视化分析结果的图形界面,用户可根据需要选择图片的保存路径和保存格式,并给所保存的图片命名。
对于不是通过Device Studio计算本征态,但含有本征态计算结果文件 EigenStates.dsf 的用户,可在如图4-1-3所示界面中选中 Simulator → Nanodcal → Analysis Plot,弹出导入本征态结果文件的图形界面,在界面中选中本征态结果文件 EigenStates.dsf,点击 打开 按钮即可弹出本征态的3D可视化分析界面。
4.1.9.2. 本征态的2D可视化分析¶
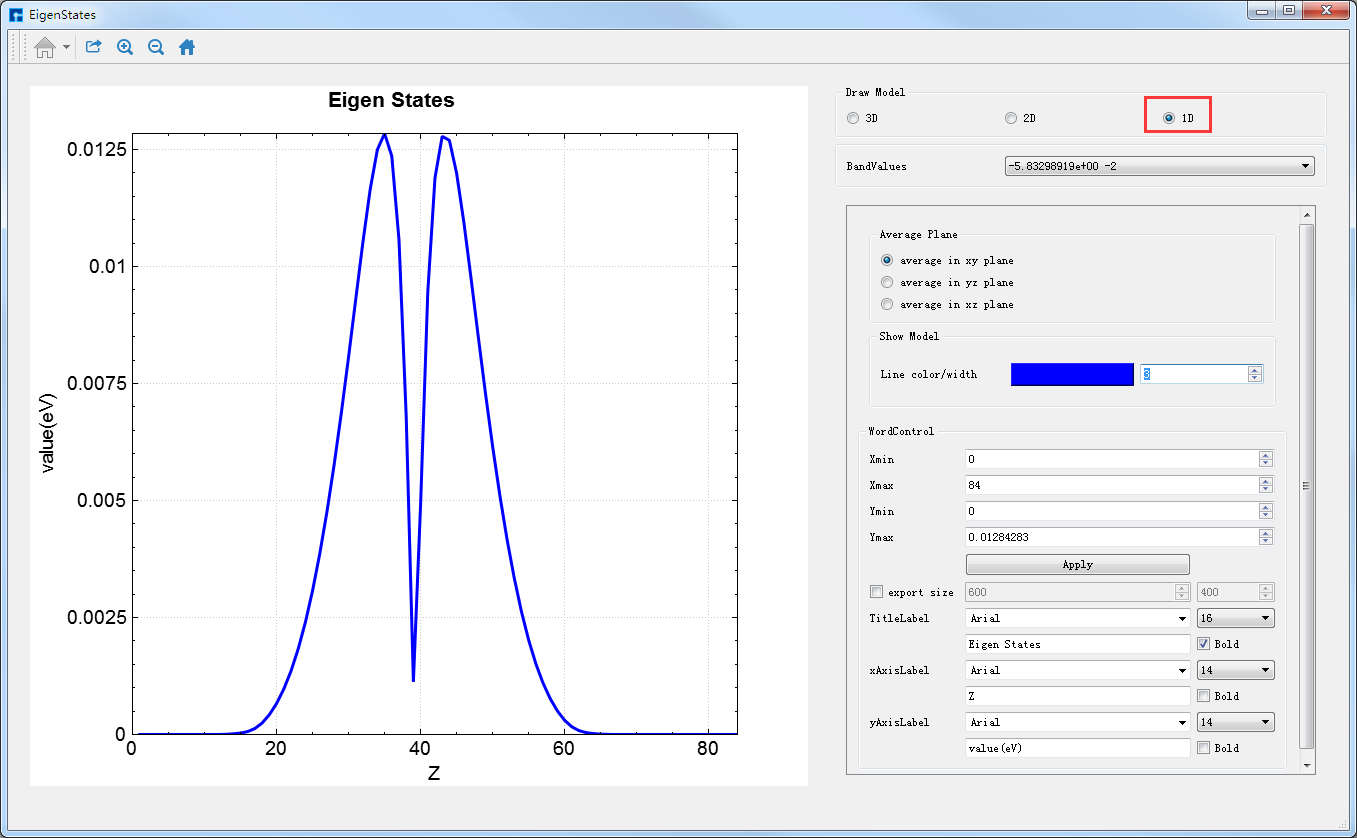
图4-1-11右侧的Draw Model栏下的三个选项3D、2D和1D分别代表着计算结果的3D、2D和1D的可视化分析。若想对Benzene分子本征态计算结果进行2D的可视化分析,直接选中Draw Model栏下的2D按钮即可切换成本征态2D的可视化分析界面如图4-1-12所示。
由图4-1-12可知,2D的可视化分析分为两种情况,一种是平面显示(Plane display),一种是立体显示(Stereoscopic display),此刻选中的按钮为Plane display,即图3-12亦为average along x的Benzene分子本征态2D平面显示的可视化分析结果。用户可根据需要通过选择Average direction栏下的3个按钮中的average along x、average along y或average along z来选择沿着x轴、y轴或z轴平均来查看并分析本征态的相关性质;可根据需要选择是否显示Colorbar,选择合适的Colormap,修改坐标轴的取值范围,修改标题和坐标轴的字体类型、字体大小以及字体是否加粗等。
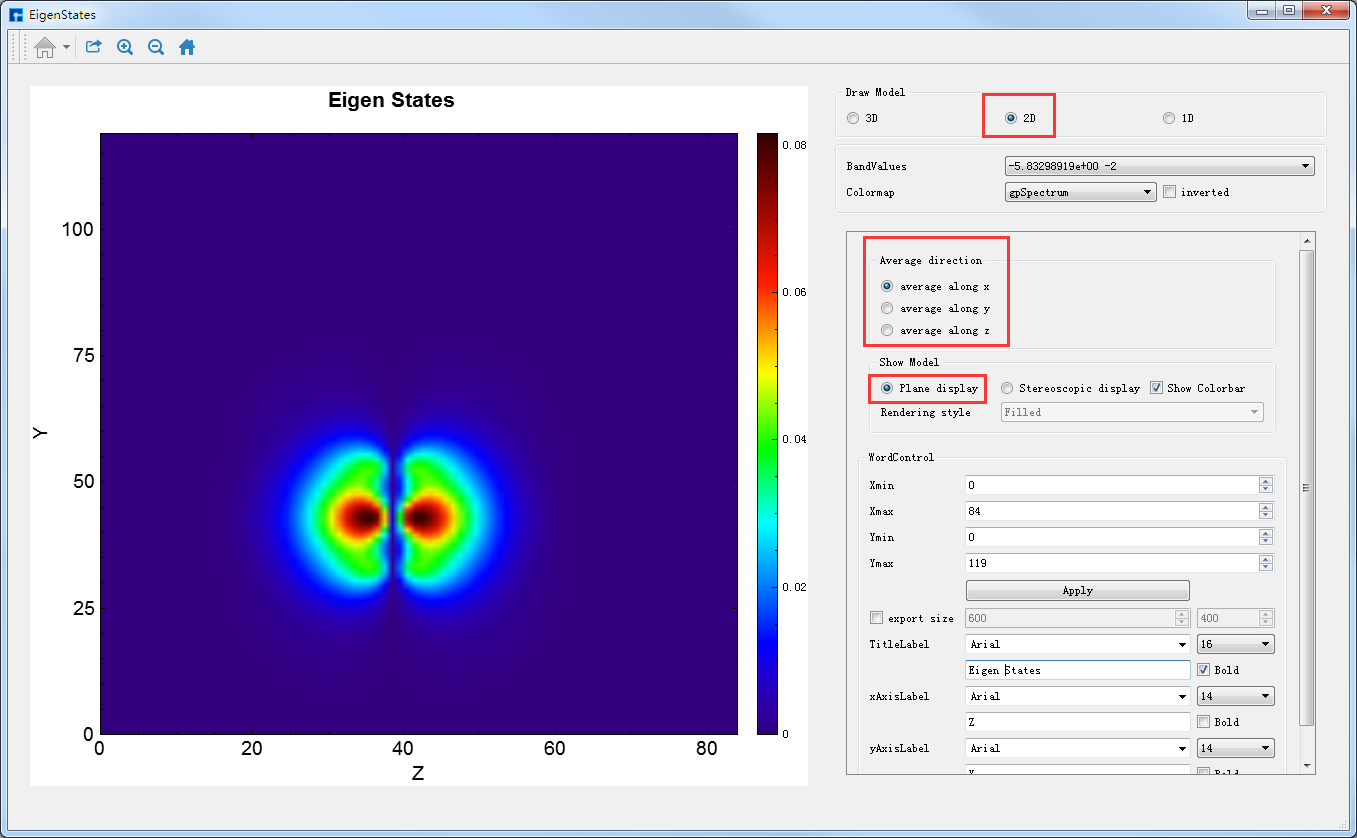
图 4-1-12 Benzene分子本征态的2D可视化分析界面(平面显示)¶
若想切换为立体显示,直接选中如图4-1-12中的Stereoscopic display按钮即可,切换后的图形界面如图4-1-13所示,图4-1-13亦为average along x的Benzene分子本征态2D立体显示的可视化分析结果。对于如图4-1-13图中左侧的图像,用户可通过滚动鼠标将图像放大或缩小;可通过拖动鼠标使图像发生任意角度和任意方向的旋转;可通过按住鼠标中键,拖动鼠标将图像进行平移。
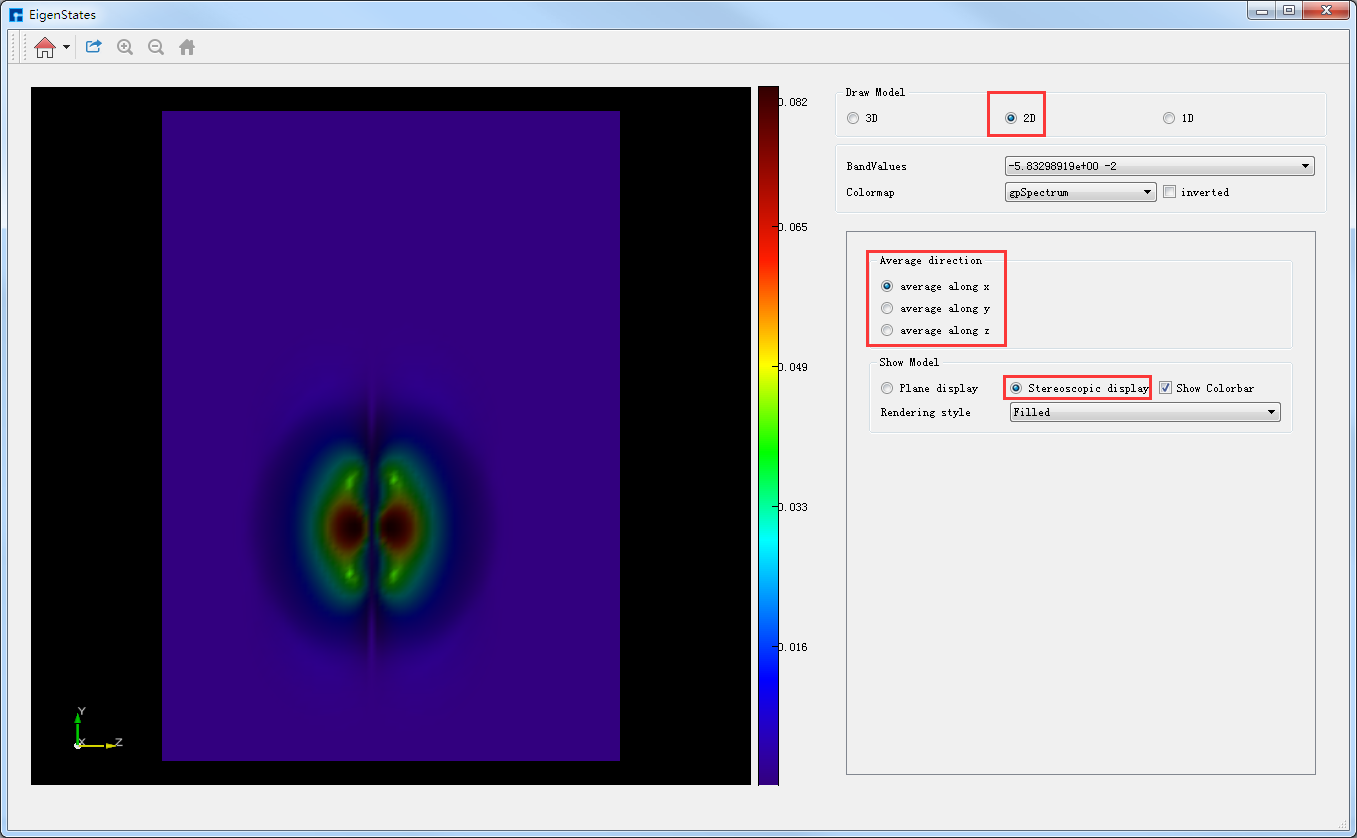
图 4-1-13 Benzene分子本征态的2D可视化分析界面(立体显示)¶
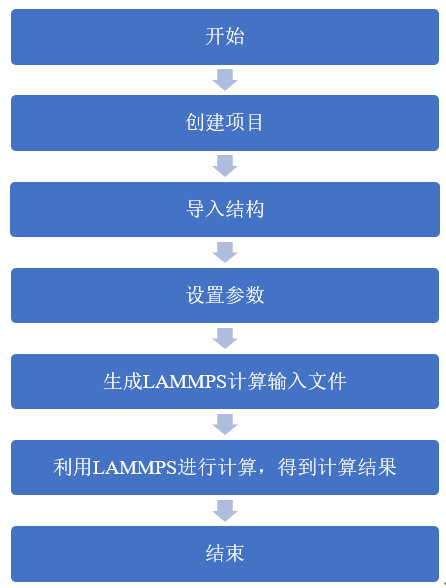
4.2. LAMMPS 实例¶
LAMMPS 即 Large-scale Atomic/Molecular Massively Parallel Simulator , 大规模原子分子并行模拟器 ,主要用于 分子动力学 相关的一些计算和模拟工作。一般来讲,分子动力学所涉及到的领域,LAMMPS代码也都涉及到了。LAMMPS由美国Sandia国家实验室开发,以GPL license发布,即开放源代码且可以免费获取使用,这意味着使用者可以根据自己的需要自行修改源代码。LAMMPS可以支持包括气态,液态或者固态相形态下、各种系综下、百万级的原子分子体系,并提供支持多种势函数,具有良好的并行扩展性。
分子动力学模拟 ( Molecular Dynamics , MD )是近年来飞速发展的一种分子模拟方法,已经被广泛应用于化学化工、材料科学与工程、物理、生物医药等科学和技术领域,起到越来越重要的作用。MD模拟用来研究不能用解析方法来解决的复合体系的平衡性质和力学性质,用来搭建理论和实验的桥梁,在数学、生物、化学、物理学、材料科学和计算机科学交叉学科占据重要地位。
LAMMPS软件安装 :详见 https://lammps.sandia.gov/doc/Install.html
4.2.2. LAMMPS创建项目¶
双击 Device Studio 图标快捷方式启动软件,根据界面提示选择创建一个新的项目 Create a new Project 或打开一个已经存在的项目 Open an existing Project 的按钮,选中之后点击界面中的 OK 按钮即可。若选择创建一个新的项目,用户可根据需要选择保存路径、给项目命名。
4.2.3. LAMMPS导入结构¶
LAMMPS中有三种定义原子的方式,从data或restart文件中读取、使用lattice, region, create_box, create_atoms命令在晶格上创建原子、使用replicate命令复制原子以生成更大的体系。简单地说,一种是依靠命令创建,一种是从其他文件导入。目前,软件在导入结构后才能使用界面设置参数。如图4-2-2,选择菜单栏 File 下的 Import ,选中结构文件并导入,支持 *.hzw 、 *.cif 、 *.xyz 、 POSCAR 等多种格式,同时在图形界面显示该结构,如图4-2-3所示。
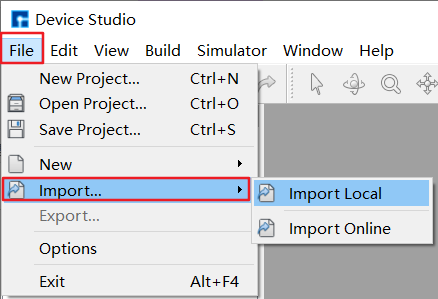
图 4-2-2: 导入结构文件菜单栏¶
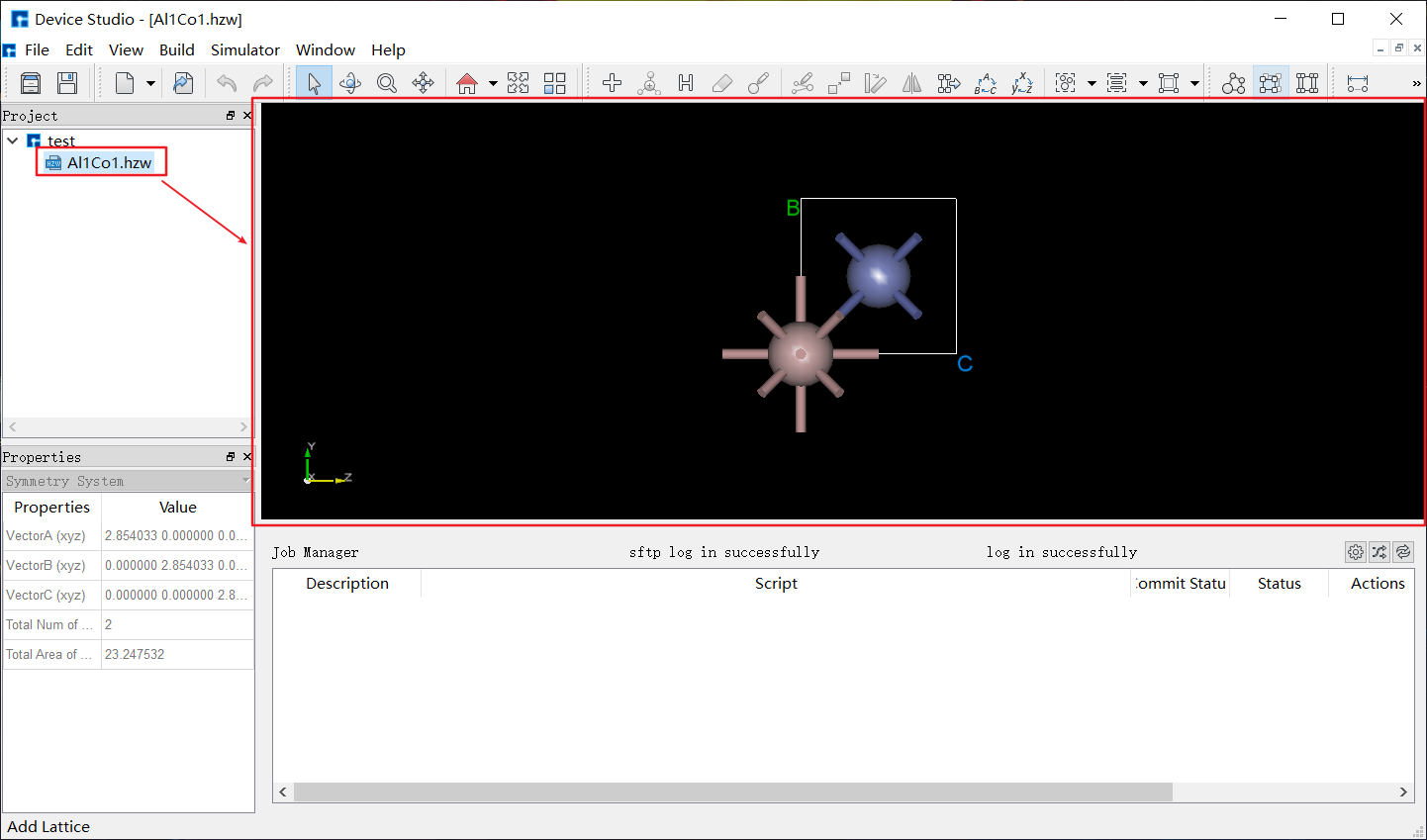
图 4-2-3: 导入结构并显示¶
4.2.4. LAMMPS参数设置¶
选择菜单栏中 Simulator → Lammps,弹出参数设置界面。
LAMMPS参数设置界面主要分为三个选项卡 Initialization (初始化)、 Simulation box (模拟盒子)和 Settings (设置)。
4.2.4.1. Initialization模块参数设置及含义¶
在 initialization 选项卡中对与初始化相关的基本参数进行修改,如图4-2-4所示,包括设置开启或关闭牛顿第三定律的 Newton 、对特定加速包设置的 Package 、后缀 Suffix 、模拟的单位 Units 、设置使用哪种类型的原子 Atom_Style 。其中, Package 、 Suffix 默认不设置,也可以勾选进行相关设置,其余参数必须进行设置。
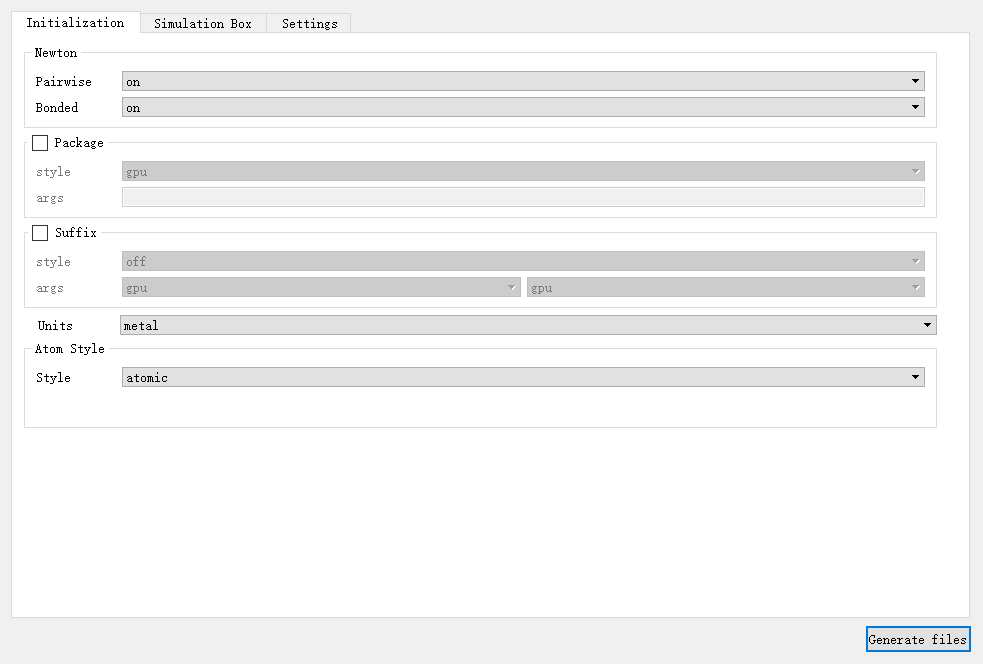
图 4-2-4: “Initialization”模块及默认值¶
4.2.4.2. Simulation box模块参数设置及含义¶
在 Simulation box 选项卡中对与模拟盒子相关的基本参数进行修改,如图4-2-5所示,包括设置三个方向边界条件的 Boundary 、模拟盒子属性的 Box 、模拟维度的 Dimension 。其中, Box 默认不设置,也可以勾选进行相关设置,其余参数必须进行设置。
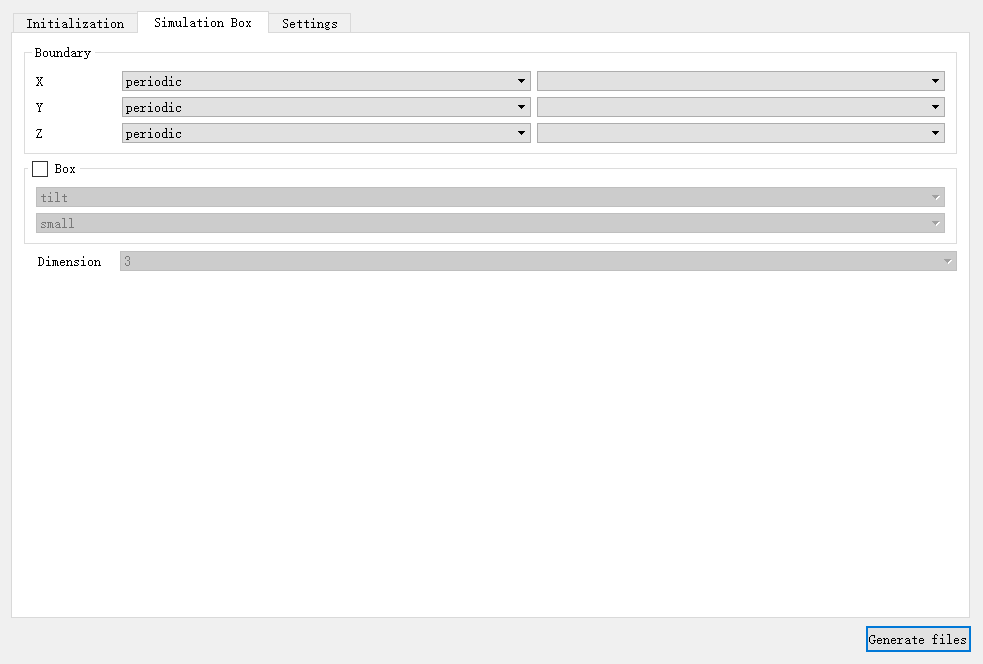
图 4-2-5: “Simulation box”模块及默认值¶
4.2.4.3. Settings模块参数设置及含义¶
由于LAMMPS在同一个输入脚本中可以采用多条相同的命令在不同的位置、进行不同参数的设置,因此完成基本参数设置后,选择 Settings 选项卡,如图4-2-6所示,分为 Commands 、 Settings 、 Parameters 三个部分。
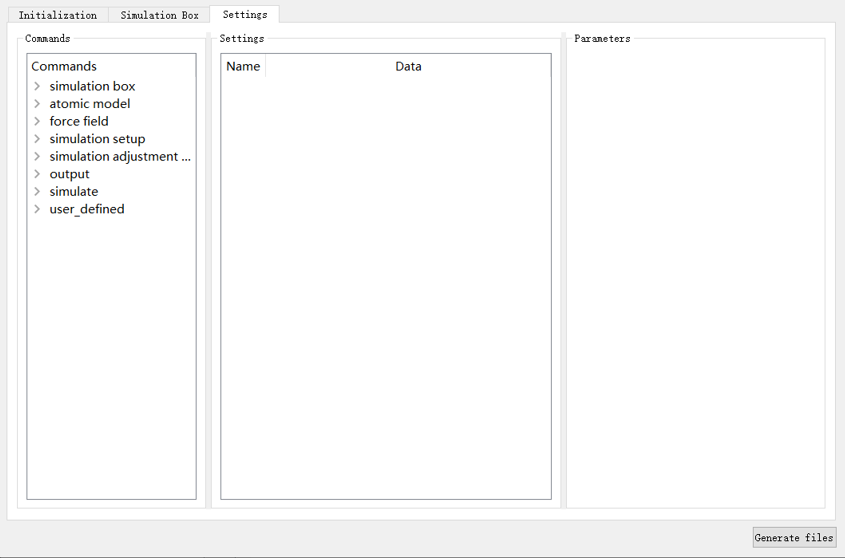
图 4-2-6: “Settings”模块¶
Commands 中包含了多条LAMMPS命令,主要分为八个部分: simulation box (仿真盒子)、 atomic model (原子模型)、 force field (力场)、 simulation setup (仿真设置)、 simulation adjustment & calculation settings (仿真调整和计算设置)、 output (输出)、 simulate (仿真)以及 user_defined (用户自定义)。
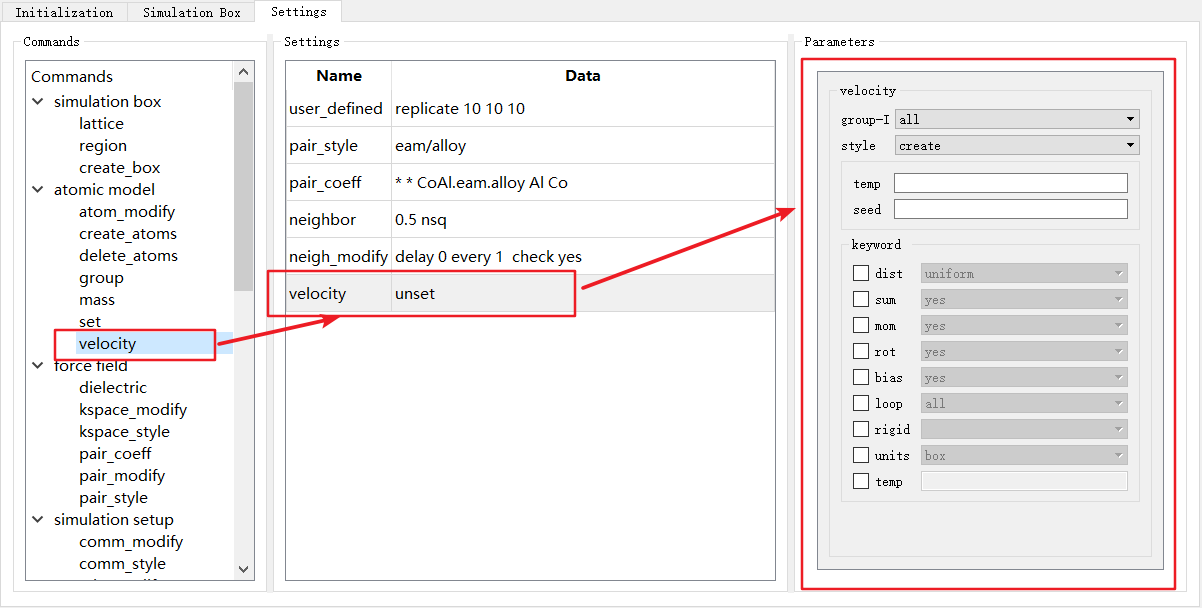
图 4-2-7: “Settings”模块中添加velocity命令¶
如图4-2-7,双击 Commands 中的命令名称如 velocity ,命令以表格中新行的形式被加入到中间 Settings 部分,也可以拖拽命令插入到 Settings 部分的任意位置。
Settings 部分分为两列, Name 显示命令的名称, Data 显示通过右侧 Parameters 设置好的参数。
如图4-2-7中 velocity unset 行,此时参数尚未进行设置,当对 Parameters 中的参数设置完成后,需要单击 Settings 中任意行,此时 Data 部分将自动填入设置好的参数,如图4-2-8。
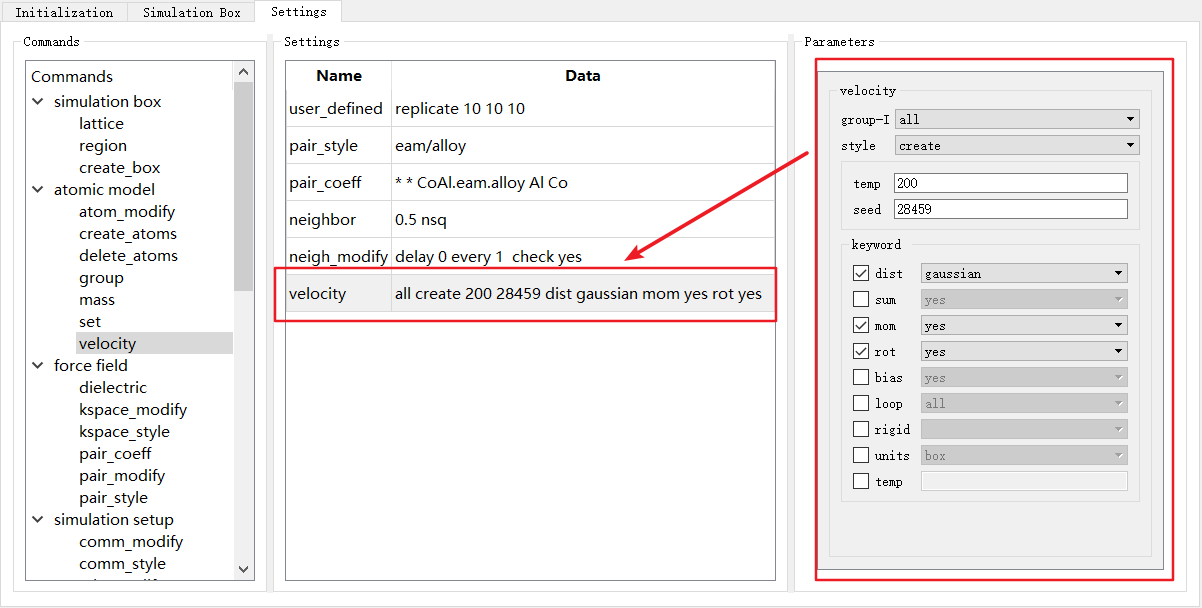
图 4-2-8: “Settings”模块中设置velocity参数¶
LAMMPS不同的命令的参数不同,因此选择不同命令时, Parameters 也对应发生变化。
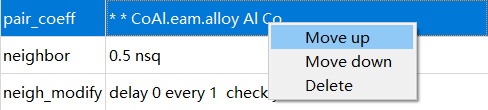
图 4-2-9: 调整命令位置或删除该条命令¶
在中间 Settings 部分,可以对每行命令进行上下拖拽调整顺序或者右键进行 Move up 、 Move down 或 Delete 操作,如图4-2-9所示。
点击已经设置好的命令行,可以随时在 Parameters 中进行参数的修改。