2. 快速入门¶
本章将介绍DS-PAW的各种功能的基本使用,具体包括: 结构弛豫计算 、 自洽计算 、 能带(投影能带)计算 、 态密度(投影态密度)计算 、 自洽中直接计算能带和态密度 、势函数计算 、 电子局域密度计算 、 部分电荷密度计算 、 杂化泛函计算 、 范德瓦尔斯修正计算 、 偶极修正计算 、 DFT+U计算 、 背景电荷计算 、 光学性质计算 、 频率计算 、 弹性常数计算 、 过渡态计算 、 声子谱计算 、 自旋轨道耦合计算 、 分子动力学模拟 、 外加电场计算 、铁电计算 、bader电荷计算 、能带反折叠计算 、 介电常数计算 、 压电张量计算 、固定基矢弛豫计算 、声子热力学性质计算 、solid state neb计算;在DS-PAW软件中大致的可以将参数分类几类:与物理结构相关的参数、与计算性质相关的参数、与计算精度相关的参数、与收敛相关的参数,这些参数中有很多都是有默认值的,且绝大多数参数不会影响计算结果; 因此本章只设置了一些特别重要的参数,同时将对这些参数进行简单的讲解。
2.1. relax结构弛豫¶
在密度泛函理论(DFT)中结构弛豫指的改变初始的结构的晶胞及原子位置从而优化得到总能量的局域最小值。 通过结构弛豫计算,可以减少在每个原子上的受力,从而得到较为稳定的结构(一定程度上,可以通过计算声子谱或者频率来验证结构的稳定性)。 使用建模软件搭建的结构一般情况下原子受力都会比较大,且即使是其他DFT软件优化过的结构,在另外一个DFT计算软件中原子受力也不一定是最小的,因此一般情况下在计算某个结构的具体性质之前需要进行结构弛豫计算。
2.1.1. \(Si\) 原子结构弛豫输入文件¶
输入文件包含参数文件 relax.in 和结构文件 structure.as , relax.in 如下:
# task type
task = relax
#system related
sys.structure = structure.as
sys.symmetry = false
sys.functional = LDA
sys.spin = none
#scf related
cal.methods = 2
cal.smearing = 1
cal.ksamping = G
cal.kpoints = [10, 10, 10]
cal.cutoffFactor = 1.5
#relax related
relax.max = 60
relax.freedom = atom
relax.convergence = 0.05
relax.methods = CG
io.outStep = 1
io.wave = false
io.charge = false
relax.in 文件大致可以分为4部分参数:
第一部分指定计算类型,计算类型中只有task一个参数:
task:设置计算类型,本次计算为relax结构弛豫;
第二部分指定系统相关参数,系统相关参数以sys.开头,一般是一些与体系的结构、泛函、磁性、对称性相关的参数:
sys.structure:指定体系的结构文件,DS-PAW支持 .as 和 .json 的结构文件格式, .as 文件可以直接使用Device Studio软件生成,也可以自己手动生成;sys.symmetry:设置DS-PAW计算时是否需要使用对称性;sys.functional:设置泛函,目前程序支持 LDA 及 PBE 泛函;sys.spin:设置体系的磁性,由于 Si 没有磁性,因此将sys.spin设置为 none ;
第三部分指定计算相关的参数,这类参数以cal.开头:
cal.methods: 设置自洽电子步优化方法,2表示使用 Residual minimization 方法;cal.smearing:设置每个波函数的部分占有数方法,1表示使用 Gaussian smearing 方法;cal.ksamping:自动生成布里渊区 k点 网格方法,G表示使用 Gamma centered 方法;cal.kpoints: 设置布里渊区 k点 网格取样大小,一般K点的大小需要根据体系晶格的大小和体系的周期性来设置,通常建议在具有周期性的方向上晶格矢量与K点的乘积不小于40,在不具有周期性的方向上将 K点 设置为1即可;
第四部分指定结构弛豫相关参数,例如结构弛豫的方法、结构弛豫的类型、结构弛豫的精度等相关参数,结构弛豫指的是优化原子位置得到总能量的局域最小值的结构,一般也称之为离子步优化;
relax.max:设置结构驰豫时,最大的离子步步数;relax.freedom:设置结构驰豫的自由度,atom表示只弛豫原子;all表示弛豫晶格常数和原子;volume表示只弛豫晶格;relax.convergence:设置结构驰豫时,原子受力的收敛判据;relax.methods:设置结构驰豫的方法,CG表示共轭梯度法;
structure.as 文件参考如下:
Total number of atoms
2
Lattice
0.00 2.75 2.75
2.75 0.00 2.75
2.75 2.75 0.00
Direct
Si -0.115000000 -0.125000000 -0.125000000
Si 0.125000000 0.125000000 0.125000000
structure.as 文件结构固定,必须严格对照每一行写入相应信息:
第一行为固定提示行
第二行为总的原子个数
第三行为固定提示行
第四至六行为晶胞信息
第七行为原子坐标的形式,可选值为 Direct 和 Cartesian (全拼且首字母必须为大写)
第八行开始写入原子坐标信息,每一行的开头必须标注坐标所描述的原子的名称
由于本案例为结构弛豫计算,因此手动的将第一个 Si原子 的X坐标从 -0.125 改为 -0.115 ,这样能够更好的体现结构弛豫的效果。
注解
如果想要固定原子,则在第7行加入Fix_x Fix_y Fix_z标签,然后在每个原子对应的位置加入F或T,F表示不固定,T表示固定。
Direct Fix_x Fix_y Fix_z
Si -0.115000000 -0.125000000 -0.125000000 F F F
Si 0.125000000 0.125000000 0.125000000 T T T
2.1.2. run程序运行¶
准备好输入文件之后,将 relax.in 和 structure.as 文件上传到安装了DS-PAW的环境上,这里将以linux环境为例子进行介绍;
在无图形界面的linux环境下运行软件的方式与在windows的环境下运行程序会有很大的区别,在linux下需要通过命令行的方式来运行程序;
一般情况下需要先加载一下环境变量,通常会将需要用的环境变量写入文本或者 .bashrc ,通过 source 的命令来加载环境;
环境加载完成之后,运行 DS-PAW relax.in ,此时使用单机版的DS-PAW进行计算;如需并行计算则需运行 DS-PAW -mpi mpirun -mpiargs "-n 2" relax.in ,-mpi 指定mpirun的名称,
-mpiargs 指定mpirun后面的参数,用双引号括起来。
如果你需要使用排队系统(例如PBS、slurm等)提交任务,只要配置完成相应的 .pbs 或 .slurm 脚本,之后使用 qsub xx.pbs 或 sbatch xx.slurm 提交任务即可。
2.1.3. analysis计算结果分析¶
根据上述的输入文件,计算完成之后将会得到 DS-PAW.log 、 relax.json 、 system.json 、 paw_tmp/relax.tmp 这4个文件:
DS-PAW.log:DS-PAW计算之后得到的日志文件;relax.json:结构弛豫完成之后的结构文件,里面保存着每一个离子步完成之后的结构文件,可以将relax.json文件导入Device Studio软件中进行显示;生成的relax.json文件也可以直接用于之后自洽计算、能带、态密度等一系列计算中,只需在计算中将 sys.structure参数 指定当前relax.json文件即可;system.json:system.json文件中保存着最后一个离子步之后的计算结果,包含结构、磁矩、能量本征值、占据态、K点信息、原子受力、stress等相应的信息;
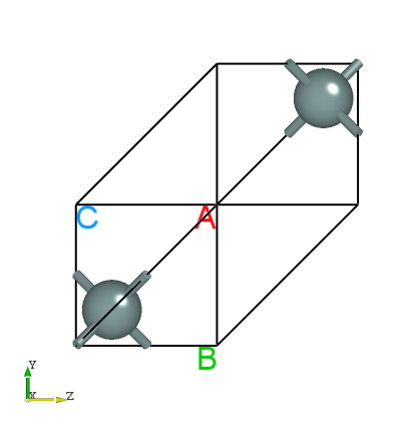
将 relax.json 拖入Device Studio查看结构如下所示:
除查看最终构型,DS可以以gif动画的形式展示弛豫过程中结构变化的过程,在面板依次选择 Simulator-->DS-PAW-->Analysis Plot-->relax.json-->Trajectory 即可,以下为截取其中一帧的图像:
使用文本编辑器或者在线显示 json格式 可打开 realx.json 文件,更简单的方法是查看paw_tmp文件夹下的 relax.tmp 文件,可得弛豫结束后的结构数据如下:
Total number of atoms
Lattice
0 2.75 2.75
2.75 0 2.75
2.75 2.75 0
Direct
Si 0.88019174 0.87481749 0.87481752
Si 0.12980826 0.12518251 0.12518248
本次结构弛豫计算进行了3个离子步的计算,弛豫结束的构型中,手动移动的第一个Si原子在x方向上的坐标在弛豫计算之后完成校正。
注解
单机版DS-PAW运行命令为软件名+输入文件名,如果你的输入文件名为abc.in那么只要执行DS-PAW abc.in即可。
该弛豫计算的收敛判据选取为原子受力,若以能量作为收敛判据,可参考 relax.convergence = 1.0e-4进行设置。
2.2. scf自洽计算¶
自洽计算能够得到特定晶体的电荷密度和波函数,有了电荷密度之后才能有计算该体系的能带、态密度等电子结构性质。 特别需要注意是:自洽与能带、态密度等电子结构性质计算是有先后顺序的,必须先进行自洽计算得到电荷密度才能进一步计算能带、态密度等电子结构性质。
2.2.1. \(Si\) 原子自洽计算之准备输入文件¶
输入文件包含参数文件 scf.in 和结构文件 structure.as , scf.in 如下:
# task type
task = scf
#system related
sys.structure = structure.as
sys.symmetry = false
sys.functional = LDA
sys.spin = none
#scf related
cal.methods = 2
cal.smearing = 1
cal.ksamping = G
cal.kpoints = [10, 10, 10]
cal.cutoffFactor = 1.5
#outputs
io.charge = true
io.wave = true
scf.in 输入参数介绍:
可以看到 scf.in 的输入文件中很多参数与结构弛豫的参数名是一致的,其设置方法也是一致的,这里只着重介绍一些前面没设置过或设置有些不同的参数:
task:本次计算为 scf 自洽计算,因此将task设置为 scf ;cal.cutoffFactor:表示截断能参数cal.cutoff的系数,在DS-PAW程序中,如果cal.cutoff参数缺失,程序将根据元素的截断能设置默认的平面波截断,cal.cutoffFactor参数就是在cal.cutoff上设置乘以一个系数;io.charge:当io.charge设置为 true 时,表示计算完成之后输出电荷密度的二进制文件rho.bin和json文件rho.json,二进制rho.bin文件用于后续的后处理计算,例如能带、态密度等,rho.json文件用于显示;io.wave:当io.wave设置为 true 时,表示计算完成之后输出波函数的二进制文件wave.bin,用户可以在后续的后处理计算中选择是否使用wave.bin开始计算;
structure.as 文件参考如下:
Total number of atoms
2
Lattice
0.00 2.75 2.75
2.75 0.00 2.75
2.75 2.75 0.00
Direct
Si -0.125000000 -0.125000000 -0.125000000
Si 0.125000000 0.125000000 0.125000000
由于本案例为自洽计算只是为了计算得到特定体系的电子结构,因此不需要手动改变原子位置;
注解
io类参数只在结构弛豫和自洽中起作用,例如io.charge在其他计算情况下将不会生成rho.bin或rho.json文件;
在结构弛豫和自洽中,还能够保存elf、potential的数据,只需要将io.elf和io.potential设置为true即可;
如果用户想要使用自己优化的结构,只需在计算中将sys.structure参数指定绝对路径或相对路径下的relax.json,也可以将relax.json文件复制到本次计算的目录中,设置sys.structure=./relax.json即可;
带自旋体系的计算案例详解第二章的NiO案例。
计算时如需给体系添加背景电荷,可直接设置sys.electron参数,该参数指定价电子的总数。
2.2.2. run程序运行¶
准备好输入文件 scf.in 和 structure.as 后,将文件上传到服务器上运行,按照结构弛豫中介绍的方法执行 DS-PAW scf.in 。
2.2.3. analysis计算结果分析¶
根据上述的输入文件,计算完成之后将会得到 DS-PAW.log 、 system.json 、 rho.bin 、 rho.json 、 wave.bin 这5个文件。
rho.bin:电荷密度的二进制文件,用于后续的后处理计算;rho.json:电荷密度的 json 格式文件,用来显示电荷密度的结果;wave.bin:波函数的二进制文件,用于后续计算;
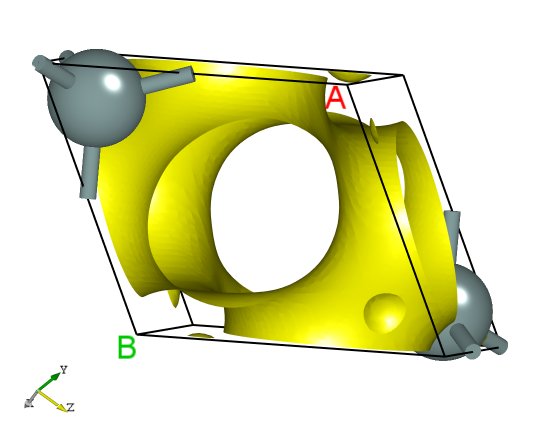
使用 Device Studio 可直接对 rho.json 文件处理出图,其操作步骤为:Simulator-->DS-PAW-->Analysis Plot,选择 rho.json 即可,可根据作图要求自定义设置面板参数,处理可得一维、二维、三维电荷密度图,其中三维图如下所示:
另可使用python脚本将 rho.json 格式的转化成 VESTA 软件支持的格式,具体操作见 辅助工具使用教程 部分。
2.3. band能带计算¶
本节将从自洽出发介绍如何使用DS-PAW计算能带和投影能带。以Si体系为例进行自洽计算(见2.2节 ),自洽完成之后准备能带计算和投影能带计算,并对能带和投影能带作图分别进行分析。
2.3.1. \(Si\) 能带计算输入文件¶
输入文件包含参数文件 band.in 、结构文件 structure.as 和上次自洽计算得到的二进制电荷密度文件 rho.bin , band.in 如下:
# task type
task = band
#system related
sys.structure = structure.as
sys.symmetry = false
sys.functional = LDA
sys.spin = none
#scf related
cal.iniCharge = ./rho.bin
cal.methods = 2
cal.smearing = 1
cal.ksamping = G
cal.kpoints = [10, 10, 10]
cal.cutoffFactor = 1.5
#band related
band.kpointsLabel= [G,X,W,K,G,L]
band.kpointsCoord= [0, 0, 0, 0.5, 0, 0.5, 0.5, 0.25, 0.75, 0.375, 0.375, 0.75, 0, 0, 0, 0.5, 0.5, 0.5]
band.kpointsNumber= [30, 30, 30, 30, 30]
band.in 输入参数介绍:
在能带计算中可以尽量保留 sys. 和 cal. 的参数到 band.in 中,之后设置能带计算特有的参数即可:
task:本次计算为band能带计算,设置task为 band ;cal.iniCharge:表示读取电荷密度二进制文件,支持绝对路径及相对路径,这里./表示当前路径下的rho.bin文件;
能带计算中新增了一部分能带相关的参数,这些参数只在能带计算中起作用:
band.kpointsLabel:表示能带计算时高对称点标签,一个band.kpointsCoord对应一个band.kpointsLabel;band.kpointsCoord:表示能带计算时高对此点的分数坐标,每三个数为一组;band.kpointsNumber:表示每相邻两个高对称点的间隔;
structure.as 文件同自洽计算。(见2.2节)
注解
若读取电荷密度文件rho.bin,cal.cutoffFactor和cal.cutoff须和之前自洽计算的设置保持一致,否则会出现格点数据不匹配的问题
band.kpointsCoord、band.kpointsLabel和band.kpointsNumber三个参数的维度需要统一
2.3.2. run程序运行¶
准备好输入文件 band.in 和 structure.as 以及 rho.bin 之后,将文件上传到服务器上运行,按照结构弛豫中介绍的方法执行 DS-PAW band.in 。
2.3.3. analysis计算结果分析¶
根据上述的输入文件,计算完成之后将会得到 DS-PAW.log 、 band.json 这2个文件。
band.json :能带计算完成之后的 json 数据文件;此时能带的数据也将会被保存在 band.json 中,具体的数据结构详见数据结构解析部分;
使用 Device Studio 可直接对 band.json 文件处理出图,其操作步骤为:Simulator-->DS-PAW-->Analysis Plot,选择 band.json 即可,可根据作图要求自定义设置面板参数,DS处理得到的能带图如下所示:
另可使用 python 进行数据处理,具体操作见 辅助工具使用教程 部分。
2.3.4. ioband自洽中直接计算能带¶
在2.2自洽计算的输入文件中加入io.band=true,之后将band相关参数写入 scf.in 中即可。输入文件如下:
# task type
task = scf
#system related
sys.structure = structure.as
sys.symmetry = false
sys.functional = LDA
sys.spin = none
#scf related
cal.methods = 2
cal.smearing = 1
cal.ksamping = G
cal.kpoints = [10, 10, 10]
cal.cutoffFactor = 1.5
#outputs
io.charge = true
io.wave = true
#band related
io.band=true
band.kpointsLabel= [G,X,W,K,G,L]
band.kpointsCoord= [0, 0, 0, 0.5, 0, 0.5, 0.5, 0.25, 0.75, 0.375, 0.375, 0.75, 0, 0, 0, 0.5, 0.5, 0.5]
band.kpointsNumber= [30, 30, 30, 30, 30]
此时计算出来的能带图与单独计算得到的能带图效果一致。
注解
只有在task=scf时io.band=true才能生效;
当io.band=true生效时,band相关的参数都将生效;
当io.band生效时,不再需要设置cal.iniCharge = ./rho.bin,此过程将在程序内部完成。
ioband.in文件中需给出两类k点,cal.kpoints给出自洽计算的k点,band.kpoints相关参数给出能带计算的k点,两部分k点缺一不可。
2.4. pband投影能带计算¶
投影能带是指在能带计算过程中将每条能带每个K点上的能量展开为每个原子及其轨道的贡献。
2.4.1. \(Si\) 投影能带计算输入文件¶
投影能带的输入文件包含参数文件 pw_band.in 结构文件 structure.as 和上次自洽计算得到的二进制电荷密度文件 rho.bin , pw_band.in 如下:
# task type
task = band
#system related
sys.structure = structure.as
sys.symmetry = false
sys.functional = LDA
sys.spin = none
#scf related
cal.iniCharge = ./rho.bin
cal.methods = 2
cal.smearing = 1
cal.ksamping = G
cal.kpoints = [10, 10, 10]
cal.cutoffFactor = 1.5
#band related
band.kpointsLabel= [G,X,W,K,G,L]
band.kpointsCoord= [0, 0, 0, 0.5, 0, 0.5, 0.5, 0.25, 0.75, 0.375, 0.375, 0.75, 0, 0, 0, 0.5, 0.5, 0.5]
band.kpointsNumber= [30, 30, 30, 30, 30]
band.project=true
pw_band.in 输入参数介绍:
pw_band.in : 与普通能带的区别在于在计算参数中设置了 band.project 参数:
band.project :表示在能带计算中打开能带投影的开关;
2.4.2. run程序运行¶
准备好输入文件 pw_band.in 和 structure.as 以及 rho.bin 之后,将文件上传到服务器上运行,按照结构弛豫中介绍的方法执行 DS-PAW pw_band.in 。
2.4.3. analysis计算结果分析¶
根据上述的输入文件,计算完成之后将会得到 DS-PAW.log 、 band.json 这2个文件。
band.json :能带计算完成之后的 json 数据文件;此时投影能带的数据也将会被保存在 band.json 中,具体的数据结构详见数据结构解析部分;
使用 Device Studio 可直接对 band.json 文件处理出图,其操作步骤为:Simulator-->DS-PAW-->Analysis Plot,选择 band.json 即可,可根据作图要求自定义设置面板参数。DS处理得到的能带图如下所示:
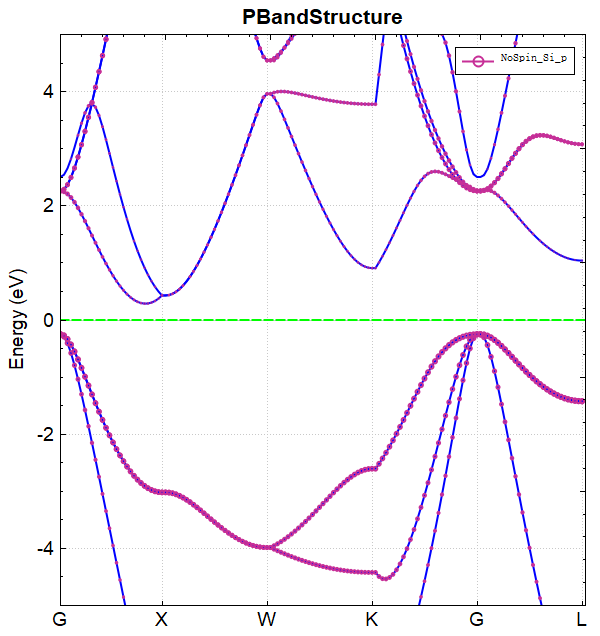
另可使用 python 进行数据处理,具体操作见 辅助工具使用教程 部分。
2.5. dos态密度计算¶
本节将从自洽出发介绍如何使用DS-PAW计算态密度计算和投影态密度。以Si体系为例进行自洽计算(见2.2节),自洽完成之后准备态密度计算和投影态密度能带计算,并对能带和投影能带作图分别进行分析。
2.5.1. \(Si\) 态密度计算输入文件¶
输入文件包含参数文件 dos.in 结构文件 structure.as 和上次自洽计算得到的二进制电荷密度文件 rho.bin , dos.in 如下:
# task type
task = dos
#system related
sys.structure = structure.as
sys.symmetry = false
sys.functional = LDA
sys.spin = none
#scf related
cal.iniCharge = ./rho.bin
cal.methods = 2
cal.smearing = 4
cal.ksamping = G
cal.kpoints = [20, 20, 20]
cal.cutoffFactor = 1.5
cal.sigma = 0.05
#dos related
#dos related
dos.range=[-10, 10]
dos.resolution=0.05
dos.EfShift = true
dos.in 输入参数介绍:
在态密度计算中可以尽量保留 sys. 和 cal. 的参数到 dos.in 中,之后设置态密度计算特有的参数即可:
task:本次计算为dos态密度计算,设置task为 dos ;cal.iniCharge:表示读取电荷密度二进制文件,支持绝对路径及相对路径,这里./表示当前路径下的rho.bin文件;cal.kpoints:为得到较为精确的态密度计算结果,需要设置更为密的 K 网格,因此需要重新设置cal.kpoints,一般该参数的取值为自洽时的两倍左右;cal.sigma:在态密度计算中为了得到比较精细的态密度,往往需要将 sigma 的值设置为尽可能的小;
态密度计算中新增了一部分态密度相关的参数,这些参数只在态密度计算中起作用:
dos.range:态密度计算时能量的区间;dos.resolution:表示态密度计算能量间隔精度,态密度计算的点数即为 dos.range 的差值与 dos.resolution 的比值+1;dos.EfShift:当dos.EfShift为 true 时,表示将 dos.range 中设置的能量是相对于自洽费米能级的相对能量;
structure.as 文件同自洽计算。(见2.2节)
注解
需要设置cal.iniCharge参数的时候,cal.cutoffFactor和cal.cutoff参数必须和之前自洽计算的结果一致,不然读取rho.bin的时候会出现格点数据不匹配的问题。
2.5.2. run程序运行¶
准备好输入文件 dos.in 和 structure.as 以及 rho.bin 之后,将文件上传到服务器上运行,按照结构弛豫中介绍的方法执行 DS-PAW dos.in 。
2.5.3. analysis计算结果分析¶
根据上述的输入文件,计算完成之后将会得到 DS-PAW.log 、 dos.json 这2个文件。
dos.json :态密度计算完成之后的json数据文件;
使用 Device Studio 可直接对 dos.json 文件处理出图,其操作步骤为:Simulator-->DS-PAW-->Analysis Plot,选择 dos.json 即可,可根据作图要求自定义设置面板参数。DS处理得到的态密度图如下所示:
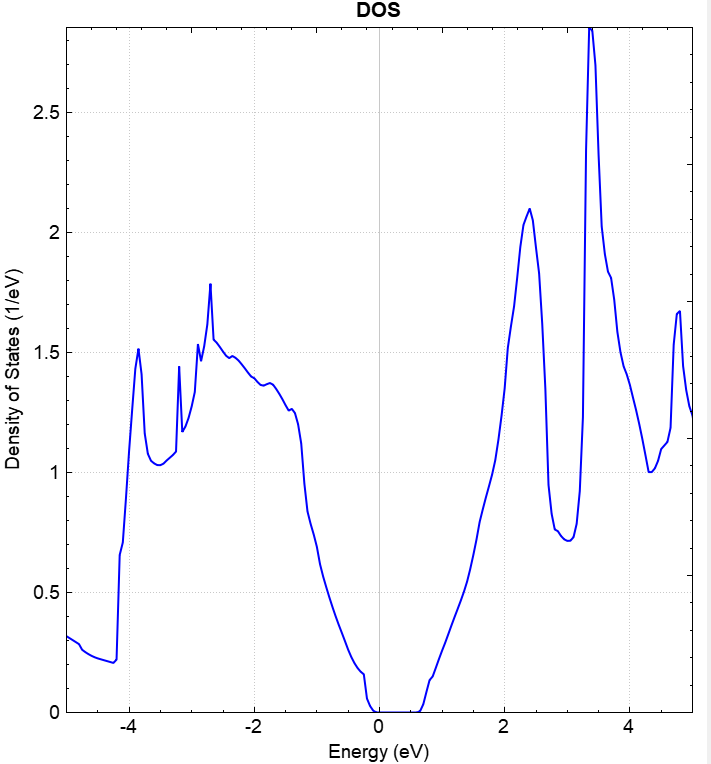
另可使用 python 进行数据处理,具体操作见 辅助工具使用教程 部分。
2.5.4. iodos自洽中直接计算态密度¶
在2.2自洽计算的输入文件中加入io.dos=true,之后将dos相关参数写入 scf.in 中即可。输入文件如下:
# task type
task = scf
#system related
sys.structure = structure.as
sys.symmetry = false
sys.functional = LDA
sys.spin = none
#scf related
cal.methods = 2
cal.smearing = 1
cal.ksamping = G
cal.kpoints = [10, 10, 10]
cal.cutoffFactor = 1.5
#outputs
io.charge = true
io.wave = true
#dos related
io.dos=true
dos.range=[-10, 10]
dos.resolution=0.05
dos.EfShift = true
注解
只有在task=scf时io.dos=true才能生效;
当io.dos=true生效时,dos相关的参数都将生效;
当io.dos生效时,不再需要设置cal.iniCharge = ./rho.bin,此过程将在程序内部完成。
iodos.in文件中cal.kpoints提供自洽计算和态密度计算的k点,两个过程的k点的大小需保持一致。
2.6. pdos投影态密度计算¶
投影态密度的计算是指在态密度计算过程中将态每个能量下的态密度展开为每个原子及其轨道的态密度贡献。
2.6.1. \(Si\) 投影态密度计算输入文件¶
投影态密度的输入文件包含参数文件 pdos.in 结构文件 structure.as 和上次自洽计算得到的二进制电荷密度文件 rho.bin , pdos.in 如下:
# task type
task = dos
#system related
sys.structure = structure.as
sys.symmetry = false
sys.functional = LDA
sys.spin = none
#scf related
cal.iniCharge = ./rho.bin
cal.methods = 2
cal.smearing = 4
cal.ksamping = G
cal.kpoints = [20, 20, 20]
cal.cutoffFactor = 1.5
cal.sigma = 0.05
#dos related
dos.range=[-10, 10]
dos.resolution=0.05
dos.EfShift = true
dos.project = true
pdos.in 输入参数介绍:
pdos.in 与普通态密度的区别在于在计算参数中设置了 dos.project 参数:
dos.project:表示在态密度计算中打开态密度投影的开关;
2.6.2. run程序运行¶
准备好输入文件 pdos.in 和 structure.as 以及 rho.bin 之后,将文件上传到服务器上运行,按照结构弛豫中介绍的方法执行 DS-PAW pdos.in 。
2.6.3. analysis计算结果分析¶
根据上述的输入文件,计算完成之后将会得到 DS-PAW.log 、 dos.json 这2个文件。
dos.json :态密度计算完成之后的 json 数据文件;此时投影态密度的数据也将会被保存在
dos.json中,具体的数据结构详见数据结构解析部分;
使用 Device Studio 可直接对 dos.json 文件处理出图,其操作步骤为:Simulator-->DS-PAW-->Analysis Plot,选择 dos.json 即可,可根据作图要求自定义设置面板参数。DS处理得到的投影态密度图如下所示:
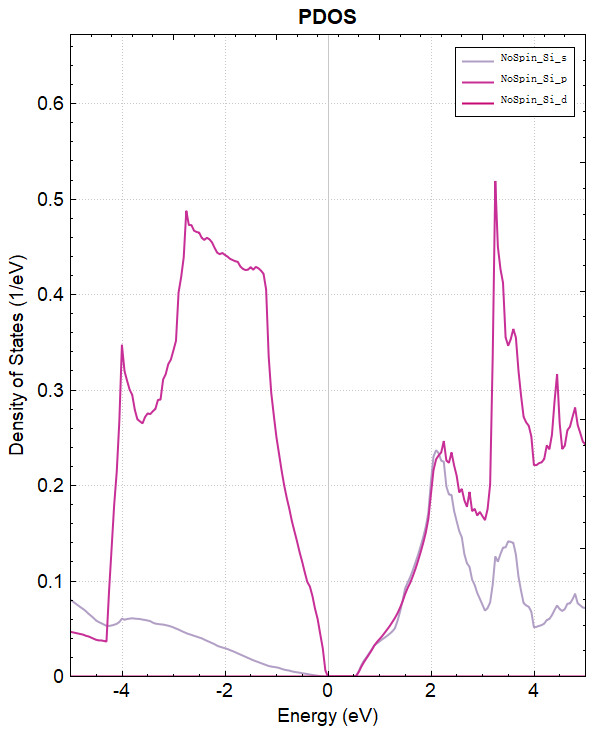
另可使用 python 进行数据处理,具体操作见 辅助工具使用教程 部分。
2.7. potential势函数计算¶
本节将从自洽出发介绍如何使用DS-PAW计算势函数。以 Si 体系为例进行自洽计算(见2.2节),自洽完成之后准备势函数输入文件,并对势函数的结果进行分析。
2.7.1. \(Si\) 势函数计算输入文件¶
输入文件包含参数文件 potential.in 结构文件 structure.as 和上次自洽计算得到的二进制电荷密度文件 rho.bin , potential.in 如下:
# task type
task = potential
#system related
sys.structure = structure.as
sys.symmetry = false
sys.functional = LDA
sys.spin = none
#scf related
cal.iniCharge = ./rho.bin
cal.methods = 2
cal.smearing = 1
cal.ksamping = G
cal.kpoints = [10, 10, 10]
cal.cutoffFactor = 1.5
#potential related
potential.type=all
potential.in 输入参数介绍:
在势函数计算中可以尽量保留 sys. 和 cal. 的参数到 potential.in 中,之后设置势函数计算特有的参数即可:
task:本次计算为potential势函数计算,设置task为 potential ;cal.iniCharge:表示读取电荷密度二进制文件,支持绝对路径及相对路径,这里./表示当前路径下的rho.bin文件;
势函数计算中新增了一个相关的参数:
potential.type:表示保存势函数的类型,当选择 all 的时候,势函数计算完成之后会同时保存静电势及总的局域势;
structure.as 文件同自洽计算。(见2.2节)
注解
需要设置cal.iniCharge参数的时候,cal.cutoffFactor和cal.cutoff参数必须和之前自洽计算的结果一致,不然读取rho.bin的时候会出现格点数据不匹配的问题;
本案例是调用自洽的结果进行势函数计算,用户也可以在task=scf时设置io.potential=true,此时也能保存potential.json文件。
如果所计算的体系需要考虑偶极修正,此时用户需要在自洽和势函数计算文件中都加入corr.dipol = true以及corr.dipolDirection参数,corr.dipol = true表示打开偶极修正的开关,corr.dipolDirection表示设置偶极修正的方向a、b、c分别表示沿着晶格矢量的a、b、c方向。
偶极修正的具体案例见应用案例:Au-Al体系功函数计算案例。
2.7.2. run程序运行¶
准备好输入文件 potential.in 和 structure.as 以及 rho.bin 之后,将文件上传到服务器上运行,按照结构弛豫中介绍的方法执行 DS-PAW potential.in 。
2.7.3. analysis计算结果分析¶
根据上述的输入文件,计算完成之后将会得到 DS-PAW.log 、 potential.json 这2个文件。
potential.json :势函数计算完成之后的 json 数据文件;
使用 Device Studio 可直接对 potential.json 文件处理出图,其操作步骤为:Simulator-->DS-PAW-->Analysis Plot,选择 potential.json 即可,可根据作图要求自定义设置面板参数作一维、二维、三维的势函数曲线。DS处理得到的势函数一维图如下所示:
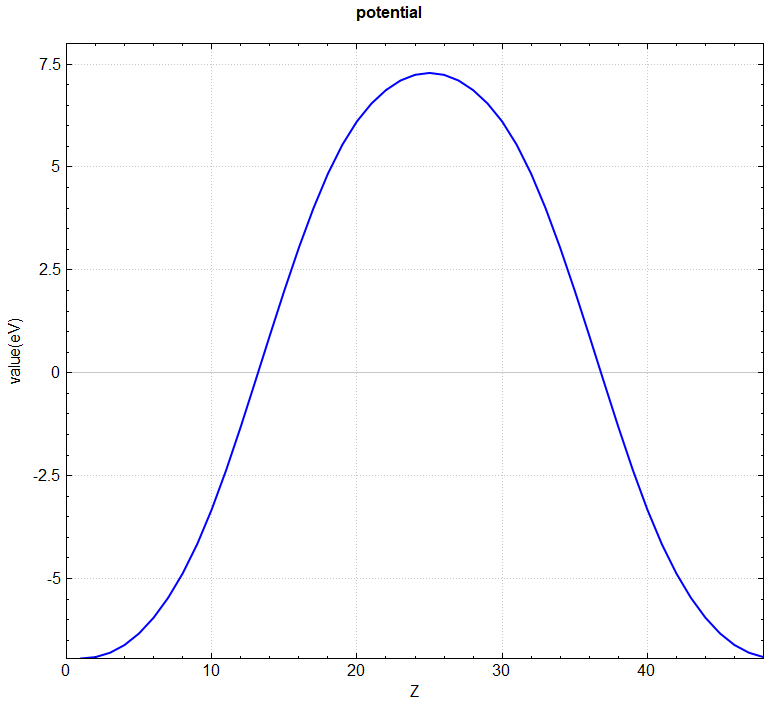
另可使用 python 脚本将 potential.json 格式的转化成 VESTA 软件支持的格式,具体操作见 辅助工具使用教程 部分。
2.8. elf电子局域密度计算¶
本节将从自洽出发介绍如何使用DS-PAW计算电子局域密度ELF。以Si体系为例进行自洽计算(见2.2节),自洽完成之后准备ELF计算,并对ELF作图进行分析。
2.8.1. \(Si\) 电子局域密度计算输入文件¶
输入文件包含参数文件 ELF.in 结构文件 structure.as 和上次自洽计算得到的二进制电荷密度文件 rho.bin , ELF.in 如下:
# task type
task = elf
#system related
sys.structure = structure.as
sys.symmetry = false
sys.functional = LDA
sys.spin = none
#scf related
cal.iniCharge = ./rho.bin
cal.methods = 2
cal.smearing = 1
cal.ksamping = G
cal.kpoints = [10, 10, 10]
cal.cutoffFactor = 1.5
ELF.in 输入参数介绍:
在ELF计算中可以尽量保留 sys. 和 cal. 的参数到 ELF.in 中:
task:本次计算为ELF计算,设置task为elf;
cal.iniCharge:表示读取电荷密度二进制文件,支持绝对路径及相对路径,这里./表示当前路径下的rho.bin文件;
structure.as 文件同(见2.2节)自洽计算的。
注解
需要设置cal.iniCharge参数的时候,cal.cutoffFactor和cal.cutoff参数必须和之前自洽计算的结果一致,不然读取rho.bin的时候会出现格点数据不匹配的问题;
本案例是调用自洽的结果进行ELF计算,用户也可以在task=scf时设置io.elf=true,此时也能保存elf.json文件。
2.8.2. run程序运行¶
准备好输入文件之后,将 ELF.in 和 structure.as 以及 rho.bin 文件上传到服务器上运行,按照结构弛豫中介绍的方法执行 DS-PAW ELF.in 。
2.8.3. analysis计算结果分析¶
根据上述的输入文件,计算完成之后将会得到 DS-PAW.log 、 elf.json 这2个文件。
elf.json :ELF计算完成之后的 json 数据文件;
使用 Device Studio 可直接对 elf.json 文件处理出图,其操作步骤为:Simulator-->DS-PAW-->Analysis Plot,选择 elf.json 即可,可根据作图要求自定义设置面板参数,得到一维、二维、三维的电子局域密度图。DS处理得到的三维电子局域密度图如下所示:
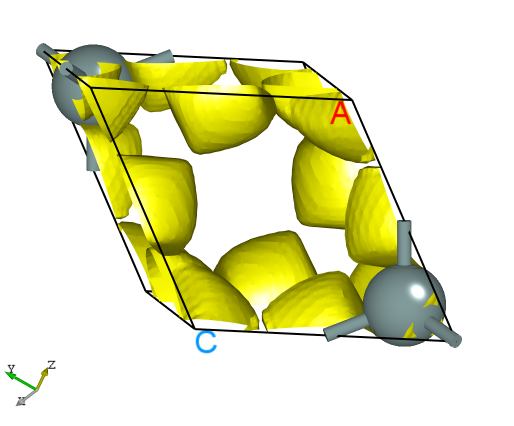
另可使用 python 脚本将 elf.json 格式的转化成 VESTA 软件支持的格式,具体操作见 辅助工具使用教程 部分。
2.9. pcharge部分电荷密度计算¶
本节将以石墨烯为例分析指定k点下特定能带的电荷密度,自洽完成之后准备部分电荷密度的计算,并对部分电荷密度作图进行分析。
2.9.1. graphene石墨烯部分电荷密度计算输入文件¶
输入文件包含参数文件 pcharge.in 和结构文件 structure.as ,上次自洽计算得到的二进制电荷密度文件 rho.bin 和二进制波函数文件 wave.bin , pcharge.in 如下:
# task type
task = pcharge
#system related
sys.structure = structure.as
sys.symmetry = false
sys.functional = PBE
sys.spin = collinear
#scf related
cal.methods = 2
cal.smearing = 1
cal.ksamping = G
cal.kpoints = [9, 9, 1]
cal.cutoffFactor = 1.5
#pcharge related
cal.iniCharge = ./rho.bin
cal.iniWave = ./wave.bin
pcharge.bandIndex = [4,5]
pcharge.kpointsIndex = [12]
pcharge.sumK= false
pcharge.in 输入参数介绍:
在部分电荷密度计算中可以尽量保留sys.和cal.的参数到 pcharge.in 中,之后设置部分电荷密度计算特有的参数即可:
task: 本次计算为部分电荷密度计算,设置task为pcharge;cal.iniCharge: 表示读取电荷密度二进制文件,支持绝对路径及相对路径,这里./表示当前路径下的rho.bin文件;cal.iniWave: 表示读取波函数二进制文件,支持绝对路径及相对路径,这里./表示当前路径下的wave.bin文件;pcharge.bandIndex: 指定需进行电荷密度分析的能带的序号,这里[4,5]表示分析能带4和能带5的电荷密度;pcharge.kpointsIndex: 指定计算某条能带的电荷密度时所用K点数,这里[12]表示分析两条能带的电荷密度时k点都取12;pcharge.sumK: 表示计算部分电荷密度之后保存数据是否将所有K点,不同能带的数据相加。这里false表示不相加;
structure.as 文件参考如下:
Total number of atoms
2
Lattice
2.46120000 0.00000000 0.00000000
-1.23060000 2.13146172 0.00000000
0.00000000 0.00000000 6.70900000
Cartesian
C 0.61530000 0.35524362 3.35450000
C 0.61530000 1.77621810 3.35450000
注解
部分电荷密度分两步完成,第二步必须读取自洽计算的电荷密度文件以及二进制波函数文件wave.bin。
2.9.2. run程序运行¶
准备好输入文件 pcharge.in 、 structure.as 以及自洽计算得到的 rho.bin 、 wave.bin 文件上传到服务器上运行,按照结构弛豫中介绍的方法执行 DS-PAW pcharge.in 。
2.9.3. analysis计算结果分析¶
根据上述的输入文件,计算完成之后将会得到 DS-PAW.log 、 pcharge.json 这2个文件。
pcharge.json :部分电荷密度计算完成之后的json数据文件,此时两条能带的电荷密度数据被保存在 pcharge.json 中,具体的数据结构详见数据结构解析部分;
使用 Device Studio 可直接对 pcharge.json 文件处理出图,其操作步骤为:Simulator-->DS-PAW-->Analysis Plot,选择 pcharge.json 即可,可根据作图要求自定义设置面板参数。DS处理得到的k点为 12 时能带 4 的电荷密度图如下所示:

另可使用 python 进行数据处理,具体操作见 辅助工具使用教程 部分。
2.10. hse杂化泛函计算¶
本节将以Si体系为例,介绍DS-PAW程序通过自洽中直接计算能带的方法计算杂化泛函能带,观察使用杂化泛函计算后能带带隙的变化。
2.10.1. \(Si\) 杂化泛函计算输入文件¶
输入文件包含参数文件 ioband.in 和结构文件 structure.as , ioband.in 如下:
# task type
task = scf
#system related
sys.structure = structure.as
sys.symmetry = true
sys.functional = PBE
sys.spin = none
#scf related
cal.methods = 1
cal.smearing = 1
cal.ksamping = G
cal.kpoints = [5, 5, 5]
cal.cutoffFactor = 1.5
#band related
io.band = true
band.kpointsCoord=[0.62500000,0.25000000,0.62500000,0.50000000,0.00000000,0.50000000,0.00000000,0.00000000,0.00000000,0.50000000,0.00000000,0.50000000,0.50000000,0.25000000,0.75000000,0.37500000,0.37500000,0.75000000,0.00000000,0.00000000,0.00000000]
band.kpointsLabel = [U,X,G,X,W,K,G]
band.kpointsNumber = [20,20,20,20,20,20]
band.project = false
#HSE related
sys.hybrid=true
sys.hybridType=HSE06
#outputs
io.charge = false
io.wave = false
ioband.in 输入参数介绍:
在杂化泛函计算中可以尽量保留sys.和cal.的参数到 ioband.in 中,之后设置杂化泛函计算特有的参数即可:
sys.hybrid:是否进行杂化泛函计算的开关,true表示引入杂化泛函计算;sys.hybridType:指定杂化泛函的类型,此例为HSE06;
structure.as 文件同自洽计算。(见2.2节)
注解
杂化泛函计算时赝势的类型必须为PBE。
若分两步计算能带,能带计算的输入文件中需要设置cal.iniWave。
若分两步计算能带,自洽计算和能带计算的输入文件中都需要打开sys.hybrid=true。
2.10.2. run程序运行¶
准备好输入文件 ioband.in 和 structure.as 上传到服务器上运行,按照结构弛豫中介绍的方法执行 DS-PAW ioband.in 。
2.10.3. analysis计算结果分析¶
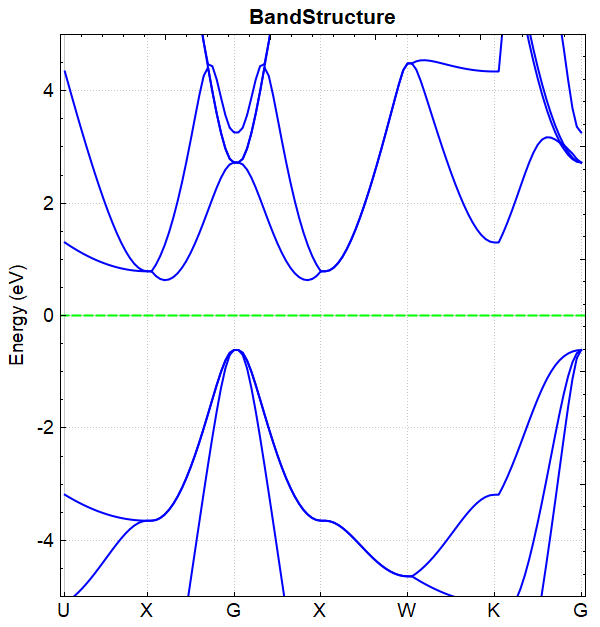
根据上述的输入文件,计算完成之后将会得到 DS-PAW.log 、 band.json 这2个文件。处理 band.json 的方法同(见2.3节)能带计算的方法,DS处理得到的 bandplot.png 文件如下所示,能带图表明打开杂化泛函计算后价带与导带之间的带隙变大,约为 1.2394 eV,不进行杂化泛函计算得到的能带带隙约为 0.6433 eV。
2.10.4. 修改杂化泛函Alpha系数¶
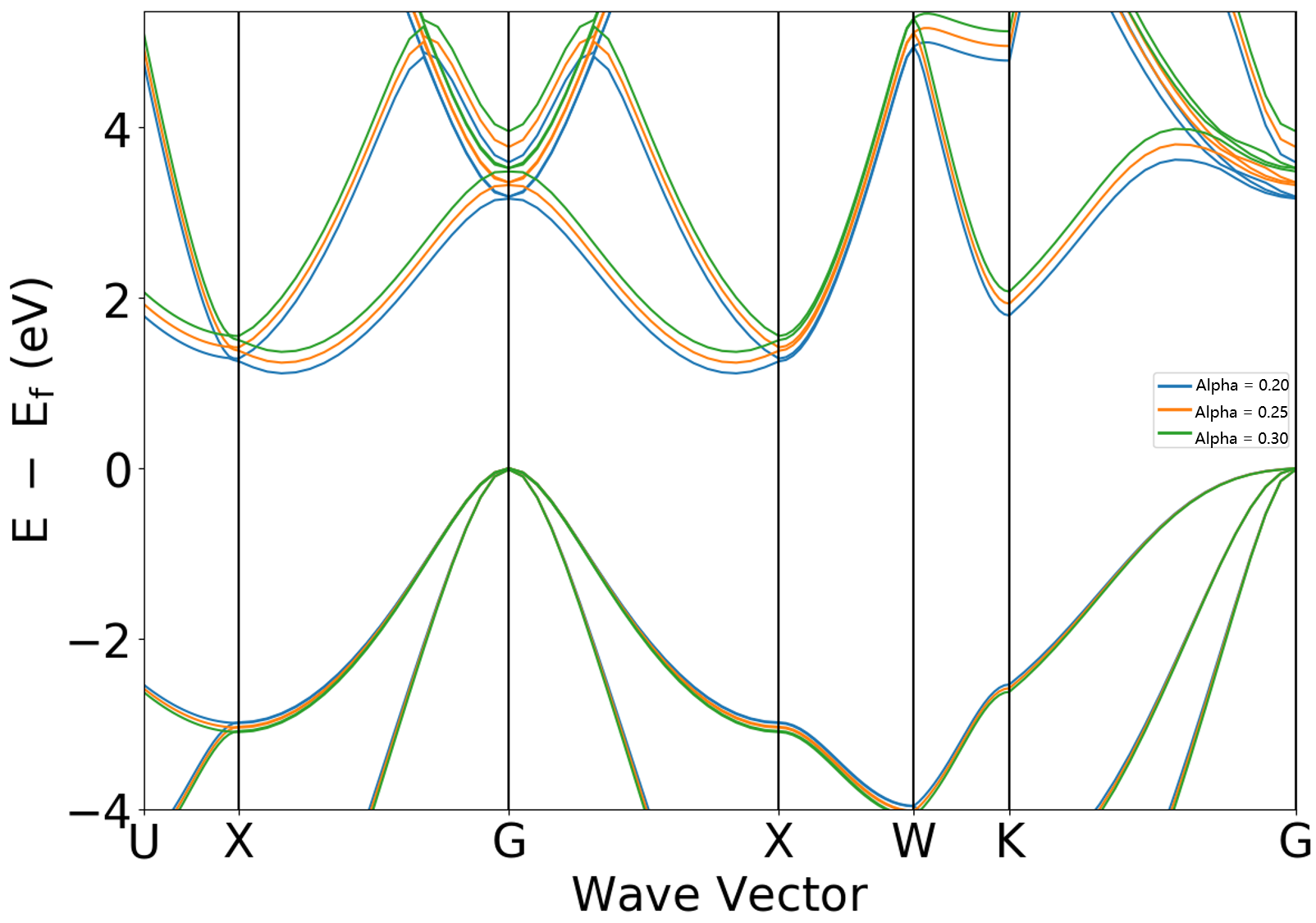
2.10.1章节展示的杂化泛函方法为HSE06,其对应的杂化泛函系数为默认值0.25,DS-PAW2022A版本新增 sys.hybridAlpha 和 sys.hybridOmega 参数用于控制杂化泛函相关系数,以 sys.hybridAlpha 为例,
若对2.10.1的计算参数做如下修改进行分别完成两次计算: scf.in 和 band.in 中增加参数 sys.hybridAlpha = 0.20,以及 scf.in 和 band.in 中增加参数 sys.hybridAlpha = 0.30 ,得到的能带对比图如下所示,分析该图可得通过加大 sys.hybridAlpha 系数可以使能带带隙进一步增大。从 band.json 文件中可读取当 sys.hybridAlpha 分别取值 0.20 、 0.25 、 0.30 时,对应带隙值分别为 1.1146 、1.2394 、 1.3665 。
2.11. vdw范德瓦尔斯修正计算¶
本节将以石墨体系的结构弛豫为例,介绍在DS-PAW中如何正确的设置范德瓦尔斯修正,并将设置范德瓦尔斯修正与不设置该参数的结果进行对比分析。
2.11.1. graphite石墨结构弛豫输入文件¶
输入文件包含参数文件 relax.in 和结构文件 structure.as , relax.in 如下:
# task type
task = relax
#system related
sys.structure = structure.as
sys.symmetry = false
sys.functional = PBE
sys.spin = none
#scf related
cal.methods = 1
cal.smearing = 1
cal.ksamping = G
cal.kpoints = [21, 21, 7]
cal.cutoff = 450
scf.convergence = 1.0e-05
#relax related
relax.max = 60
relax.freedom = all
relax.convergence = 0.01
relax.methods = CG
#vdw related
corr.VDW = true
corr.VDWType = D3G
relax.in 输入参数介绍:
在范德瓦尔斯修正计算中可以尽量保留sys.和cal.的参数到 relax.in 中,之后设置范德瓦尔斯修正计算特有的参数即可:
corr.VDW: 表示是否引入范德瓦尔斯修正,这里true表示已打开;corr.VDWType: 表示使用哪种范德瓦尔斯修正,D3G表示DFT-D3 of Grimme方法;
structure.as 文件参考如下:
Total number of atoms
4
Lattice
2.46729136 0.00000000 0.00000000
-1.23364568 2.13673699 0.00000000
0.00000000 0.00000000 7.80307245
Cartesian
C 0.00000000 0.00000000 1.95076811
C 0.00000000 0.00000000 5.85230434
C 0.00000000 1.42449201 1.95076811
C 1.23364689 0.71224492 5.85230434
注解
设置范德瓦尔斯修正时赝势的类型必须为PBE。
2.11.2. run程序运行¶
准备好输入文件之后,将 relax.in 和 structure.as 文件上传到服务器上运行,按照结构弛豫中介绍的方法执行 DS-PAW relax.in 。
2.11.3. analysis计算结果分析¶
根据上述的输入文件,计算完成之后将会得到 DS-PAW.log 、 relax.json 和 system.json 这3个文件。(为作对比另添加一组不考虑范德瓦尔斯的计算)
将 relax.json 拖入Device Studio查看结构,或直接查看 paw_tmp 目录下的relax.tmp 文件,可得弛豫结束后晶胞常数如下表所示,通过对比可发现添加范德瓦尔斯修正进行结构弛豫所得晶胞向量 c 的值与实验报道结果[1]更接近。
Procedure
a (Å)
c (Å)
vdw-D3G this work
2.464
7.083
PBE this work
2.464
7.914
Experiment
2.462
6.707
2.12. optical光学性质计算¶
本节将以Si体系为例,介绍在DS-PAW中如何进行光学性质的计算,并对一系列光学性质的物理量进行作图分析。
2.12.1. \(Si\) 光学性质计算输入文件¶
输入文件包含参数文件 scf.in 和结构文件 structure.as , scf.in 如下:
# task type
task = scf
#system related
sys.structure = structure.as
sys.symmetry = true
sys.functional = PBE
sys.spin = none
#scf related
cal.methods = 1
cal.smearing = 1
cal.ksamping = G
cal.kpoints = [4, 4, 4]
cal.cutoffFactor = 1.5
#optical related
io.optical = true
scf.in 输入参数介绍:
在光学性质计算中可以尽量保留sys.和cal.的参数到 scf.in 中,之后设置光学性质计算特有的参数即可:
io.optical: 表示是否进行光学性质的计算,当io.optical=true时,对体系进行光学性质的计算;
structure.as 文件同自洽计算。(见2.2节)
2.12.2. run程序运行¶
准备好输入文件之后,将 scf.in 和 structure.as 文件上传到服务器上运行,按照结构弛豫中介绍的方法执行 DS-PAW scf.in 。
2.12.3. analysis计算结果分析¶
根据上述的输入文件,计算完成之后将会得到 DS-PAW.log 、 optical.json 这2个文件。
optical.json :光学性质计算完成之后的 json 数据文件;此时吸光系数,折射率等物理数据被保存在 optical.json 中,具体的数据结构详见数据结构解析部分;
使用 Device Studio 可直接对 optical.json 文件处理出图,其操作步骤为:Simulator-->DS-PAW-->Analysis Plot,选择 optical.json 即可,可根据作图要求自定义设置面板参数。DS处理得到的吸光系数、消光系数、反射率、折射因子随能量的变化曲线分别如下所示:
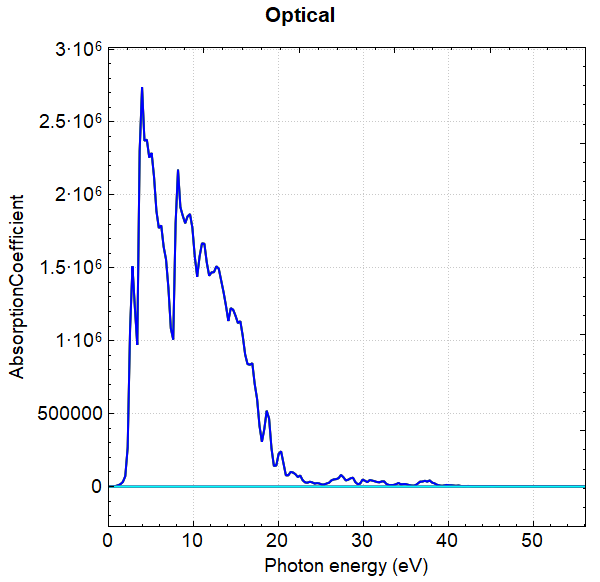
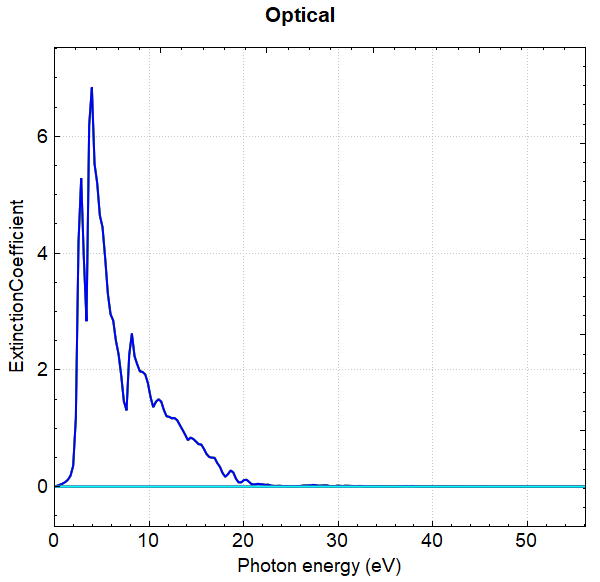
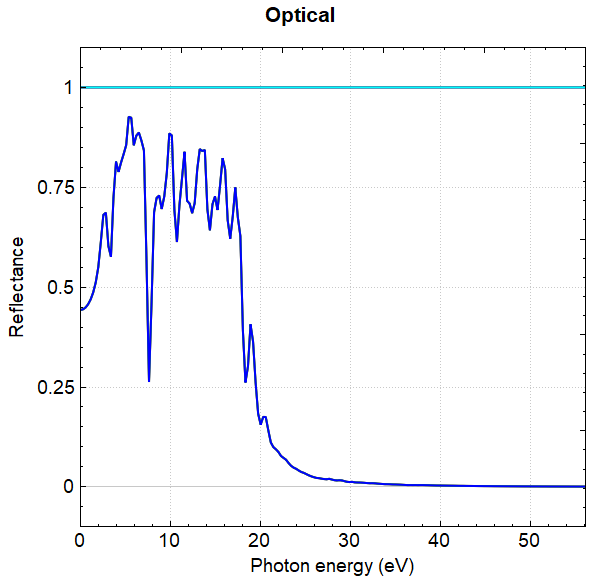
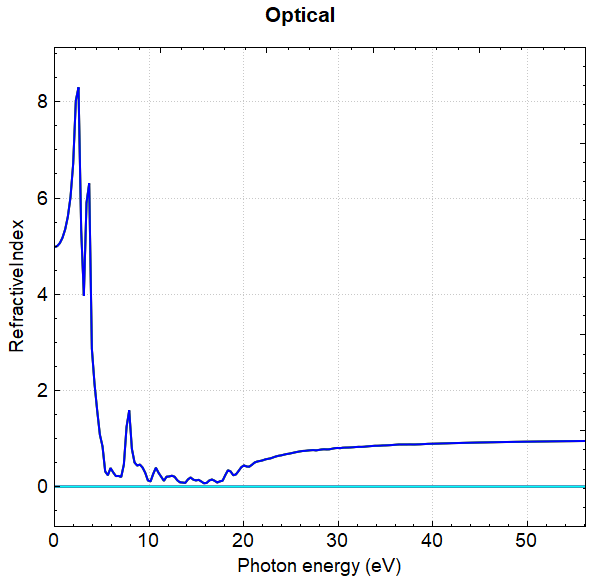
另可使用 python 进行数据处理,具体操作见 辅助工具使用教程 部分。
2.13. frequency频率计算¶
本节将以CO分子为例,介绍在DS-PAW中如何进行频率计算。
2.13.1. \(CO\) 频率计算输入文件¶
输入文件包含参数文件 frequency.in 和结构文件 structure.as , frequency.in 如下:
# task type
task = frequency
#system related
sys.structure = structure.as
sys.symmetry = false
sys.functional = PBE
sys.spin = none
#scf related
cal.methods = 2
cal.smearing = 1
cal.ksamping = MP
cal.kpoints = [9, 9, 1]
cal.cutoffFactor = 1.5
scf.convergence = 1.0e-6
#frequency related
frequency.dispOrder = 1
frequency.dispRange = 0.02
#outputs
io.charge = false
io.wave = false
frequency.in 输入参数介绍:
在频率计算中可以尽量保留sys.和cal.的参数到 frequency.in 中,之后设置频率计算特有的参数即可:
task: 本次计算为频率计算,设置task为frequency;frequency.dispOrder: 表示频率计算时原子振动的方式。1对应中心差分法,即2种原子振动方式:每个笛卡尔方向上原子的位移为±frequency.dispRange;2对应4种原子振动方式:每个笛卡尔方向上原子的位移为±frequency.dispRange和±2*frequency.dispRange;frequency.dispRange: 表示频率计算时的原子位移大小;
structure.as 文件参考如下:
Total number of atoms
2
Lattice
8.0 0.0 0.0
0.0 8.0 0.0
0.0 0.0 8.0
Cartesian Fix_x Fix_y Fix_z
O 0 0 0 T T F
C 0 0 1.143 T T F
注解
频率计算时应提高自洽计算的收敛精度,建议设置为1.0e-6以上。
CO只在z方向上两个原子可动。
2.13.2. run程序运行¶
准备好输入文件之后,将 frequency.in 和 structure.as 文件上传到服务器上运行,按照结构弛豫中介绍的方法执行 DS-PAW frequency.in 。
2.13.3. analysis计算结果分析¶
根据上述的输入文件,计算完成之后将会得到 DS-PAW.log 、 frequency.json 、 frequency.txt 这3个文件。
frequency.json :频率计算完成之后的json数据文件,此时频率数据被保存在该文件中,具体的数据结构详见数据结构解析部分。
frequency.txt :频率计算完成之后的 txt 文本文件,该文件写入频率相关数据,与 frequency.json 文件数据一致,便于用户快速获取信息。
从 frequency.txt 中可获取以下数据:
Frequency |
THz |
2PiTHz |
cm-1 |
meV |
1 f |
63.843104 |
401.138041 |
2129.576594 |
264.033942 |
2 f/i |
0.051123 |
0.321218 |
1.705293 |
0.211429 |
CO只在z方向上两个原子可动,因此只有两个频率,通过上表可以看到一个振动模式的频率约为 63.8 THz,还有一个在 0 附近的虚频,一般情况虚频小于2THz基本可以忽略不计。
2.14. elastic弹性常数计算¶
本节将以Si体系为例,介绍在DS-PAW中如何进行弹性计算。
2.14.1. \(Si\) 弹性常数计算输入文件¶
输入文件包含参数文件 elastic.in 和结构文件 structure.as , elastic.in 如下:
# task type
task = elastic
#system related
sys.structure = structure.as
sys.symmetry = true
sys.functional = PBE
sys.spin = none
#scf related
cal.methods = 1
cal.smearing = 1
cal.ksamping = G
cal.kpoints = [5, 5, 5]
cal.cutoffFactor = 1.5
scf.convergence = 1.0e-6
#frequency related
elastic.dispOrder = 1
elastic.dispRange = 0.01
#outputs
io.charge = false
io.wave = false
elastic.in 输入参数介绍:
在弹性计算中可以尽量保留sys.和cal.的参数到 elastic.in 中,之后设置弹性计算特有的参数即可:
task: 本次计算为弹性计算,设置task为elastic;elastic.dispOrder: 表示弹性计算时原子振动的方式, 1对应中心差分法;elastic.dispRange: 表示弹性计算时的原子位移大小;
structure.as 文件参考如下:
Total number of atoms
8
Lattice
5.43070000 0.00000000 0.00000000
0.00000000 5.43070000 0.00000000
0.00000000 0.00000000 5.43070000
Cartesian
Si 0.67883750 0.67883750 0.67883750
Si 3.39418750 3.39418750 0.67883750
Si 3.39418750 0.67883750 3.39418750
Si 0.67883750 3.39418750 3.39418750
Si 2.03651250 2.03651250 2.03651250
Si 4.75186250 4.75186250 2.03651250
Si 4.75186250 2.03651250 4.75186250
Si 2.03651250 4.75186250 4.75186250
注解
弹性计算时应提高自洽计算的收敛精度,建议设置在1.0e-6以上。
弹性计算不支持固定原子。
2.14.2. run程序运行¶
准备好输入文件之后,将 elastic.in 和 structure.as 文件上传到服务器上运行,按照结构弛豫中介绍的方法执行 DS-PAW elastic.in 。
2.14.3. analysis计算结果分析¶
根据上述的输入文件,计算完成之后将会得到 DS-PAW.log 、 elastic.json 、 elastic.txt 这3个文件。
elastic.json : 弹性计算完成之后的 json 数据文件,此时弹性模量被保存在 elastic.json 中,具体的数据结构详见数据结构解析部分;
elastic.txt : 弹性计算完成之后的 txt 文本文件,该文件写入弹性相关数据,与 elastic.json 文件数据一致,便于用户快速获取信息。
从 elastic.txt 文件可得如下弹性常数矩阵:
158.7644
62.9858
62.9858
0.0000
-0.0000
0.0000
62.9858
158.7644
62.9858
0.0000
0.0000
0.0000
62.9858
62.9858
158.7644
-0.0000
0.0000
0.0000
0.0000
0.0000
-0.0000
75.8807
-0.0000
0.0000
-0.0000
0.0000
0.0000
-0.0000
75.8807
-0.0000
0.0000
0.0000
0.0000
0.0000
-0.0000
75.8807
刚性弹性矩阵
0.0081
-0.0023
-0.0023
-0.0000
0.0000
-0.0000
-0.0023
0.0081
-0.0023
-0.0000
-0.0000
0.0000
-0.0023
-0.0023
0.0081
0.0000
-0.0000
0.0000
-0.0000
-0.0000
0.0000
0.0132
0.0000
-0.0000
0.0000
-0.0000
-0.0000
0.0000
0.0132
0.0000
-0.0000
0.0000
0.0000
-0.0000
0.0000
0.0132
柔性弹性矩阵
Properties
Vogit
Reuss
Hill
BulkModulus(GPa)
94.9120
94.9120
94.9120
ShearModulus(GPa)
64.6841
61.5016
63.0929
YoungModulus(GPa)
158.1297
151.7315
154.9452
PoissonRatio
0.2223
0.2336
0.2279
体积模量、剪切模量、杨氏模量和泊松比
Si体系为Cubic晶系,该晶系的独立矩阵元有三个:C11 , C12 , C44 ,分别对应表中的 158.7644、62.9858、75.8807。
2.15. neb过渡态计算¶
本节将以Pt原子体系为例,介绍在DS-PAW中如何进行过渡态计算(Cl-NEB),并对结果进行作图分析。
2.15.1. \(Pt\) 过渡态计算输入文件¶
输入文件包含参数文件 neb.in 和多个结构文件 structure##.as , neb.in 文件如下:
# task type
task = neb
#system related
sys.structure = structure.as
sys.symmetry = false
sys.functional = PBE
sys.spin = none
#scf related
cal.methods = 1
cal.smearing = 1
cal.ksamping = G
cal.kpoints = [3, 3, 1]
cal.cutoffFactor = 1.5
#neb related
neb.max = 60
neb.iniFin=true
neb.springK = 5
neb.images = 5
neb.method = LBFGS
neb.convergence = 0.1
#outputs
io.charge = false
io.wave = false
neb.in 输入参数介绍:
在过渡态计算中可以尽量保留sys.和cal.的参数到 neb.in 中,之后设置过渡态计算特有的参数即可:
task:本次计算为过渡态计算,设置task为neb;neb.stepRange: 表示过渡态计算中结构弛豫的步长;neb.max: 表示过渡态计算中结构弛豫的最大步数;neb.iniFin: 表示过渡态计算中初态结构和末态结构是否进行自洽计算,true表示进行自洽计算;neb.springK: 表示过渡态计算中弹簧系数K;neb.images: 表示过渡态计算中的中间结构的数目;neb.method: 表示过渡态计算中使用的算法;neb.convergence: 表示过渡态计算中受力的收敛标准;
structure.as 需提供多个,初态结构 structure00.as 参考如下
Total number of atoms
13
Lattice
5.6058 0 0
0 5.6058 0
0 0 16.8174
Cartesian Fix_x Fix_y Fix_z
Pt 1.40145 1.40145 1.98193 T T T
Pt 4.20435 1.40145 1.98193 T T T
Pt 1.40145 4.20435 1.98193 T T T
Pt 4.20435 4.20435 1.98193 T T T
Pt 0 0 3.89338 F F F
Pt 0 2.8029 3.89869 F F F
Pt 2.8029 0 3.96355 F F F
Pt 2.8029 2.8029 3.89338 F F F
Pt 1.43659 1.36631 5.86749 F F F
Pt 4.16921 1.36631 5.86749 F F F
Pt 1.43659 4.23949 5.86749 F F F
Pt 4.16921 4.23949 5.86749 F F F
Pt 0 2.8029 7.51195 F F F
末态结构 structure06.as 参考如下
Total number of atoms
13
Lattice
5.6058 0 0
0 5.6058 0
0 0 16.8174
Cartesian Fix_x Fix_y Fix_z
Pt 1.40145 1.40145 1.98193 T T T
Pt 4.20435 1.40145 1.98193 T T T
Pt 1.40145 4.20435 1.98193 T T T
Pt 4.20435 4.20435 1.98193 T T T
Pt 0 0 3.89338 F F F
Pt 0 2.8029 3.89869 F F F
Pt 2.8029 0 3.96355 F F F
Pt 2.8029 2.8029 3.89338 F F F
Pt 2.8029 0 7.51195 F F F
Pt 4.23949 1.43659 5.86749 F F F
Pt 1.36631 4.16921 5.86749 F F F
Pt 4.23949 4.16921 5.86749 F F F
Pt 1.36631 1.43659 5.86749 F F F
注解
过渡态计算时需准备多个结构文件,命名规则为structure##.as,##为结构序号,从00开始编号,00对应初态,依次递增。
中间结构的生成可参考"辅助工具使用教程-过渡态部分“。
过渡态计算时的结构文件structure##.as需存放在命名为##的文件夹中,文件夹序号与结构文件的序号需一致。文件夹外放置一个neb.in文件即可,在neb.in所在目录执行程序。
过渡态计算执行程序时调用的核数设置为images的整数倍。
neb计算时初态末态需先进行结构弛豫。
2.15.2. run程序运行¶
准备好输入文件之后,将 neb.in 文件和包含 structure##.as 文件的多个文件夹文件上传到服务器上运行,按照结构弛豫中介绍的方法执行 DS-PAW neb.in 。
2.15.3. analysis计算结果分析¶
根据上述的输入文件,计算完成之后初态和末态结构所在文件夹会生成自洽计算所得的 DS-PAW.log 、 system.json 这2个文件,
中间结构 structure##.as 所在文件夹 ## (参与过渡态计算的结构,由 neb.images 参数决定)会生成结构优化所得的 relax.json 、 system.json 、 neb##.json 、 paw_tmp/neb.tmp 这4个文件。
最外层目录将会生成 DS-PAW.log 、 neb.json 这2个文件,其中 neb.json 为各 neb##.json 文件的信息汇总。
neb.json :过渡态计算完成之后的 json 数据文件;此时反应坐标及能量变化等数据被保存在 neb.json 中,具体的数据结构详见数据结构解析部分;
paw_tmp/neb.tmp :中间构型在优化过程中的轨迹文件,默认neb计算过程中每个离子步记录一次结构信息;
使用 Device Studio 可直接对 neb.json 文件处理出图,其操作步骤为:Simulator-->DS-PAW-->Analysis Plot,选择 neb.json 即可,可根据作图要求自定义设置面板参数。DS处理得到的势垒曲线及image3在弛豫过程中的能量与受力曲线如下所示:
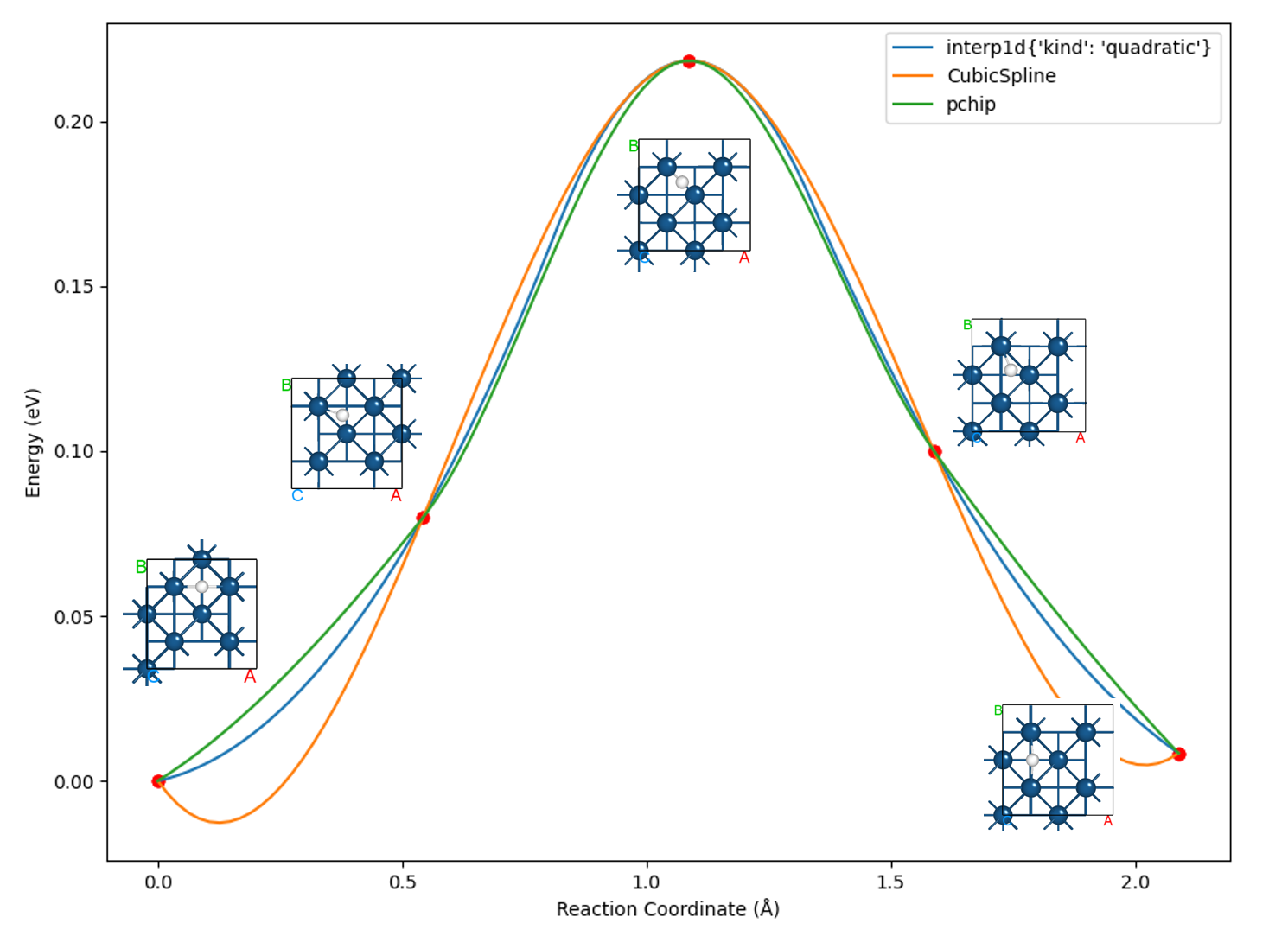
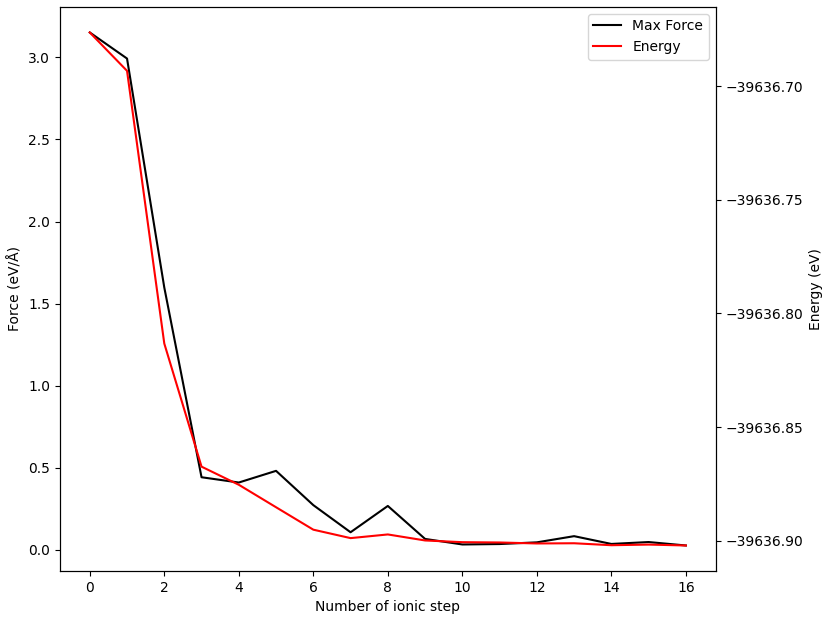
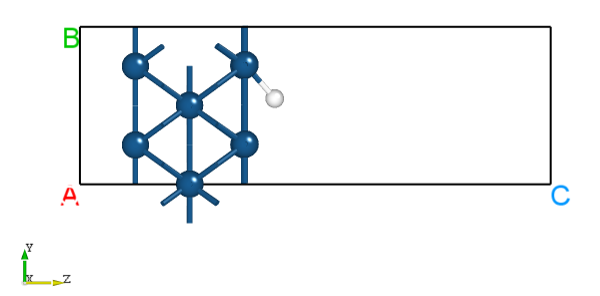
除此以外,DS可以以gif动画的形式展示过渡态搜寻中的轨迹变化,在面板依次选择 Simulator-->DS-PAW-->Analysis Plot-->neb.json-->Trajectory 即可,以下为截取其中一帧的图像:
另可使用 python 进行数据处理,具体操作见 辅助工具使用教程 部分。
2.16. phonon声子谱计算¶
本节介绍DS-PAW程序如何进行声子计算及声子能带和声子态密度计算。DS-PAW支持两种声子谱计算的方法:fd有限位移法和dfpt密度泛函微扰理论方法。本节以单个MgO体系为例,介绍如何用dfpt方法计算声子能带和态密度,并对声子能带和态密度作图进行分析。
2.16.1. \(MgO\) 声子谱能带计算输入文件¶
输入文件包含参数文件 phonon.in 和结构文件 structure.as , phonon.in 如下:
task = phonon
sys.structure = structure.as
sys.functional = PBE
sys.spin = none
cal.methods = 1
cal.smearing = 1
sys.symmetry = true
scf.convergence = 1.0e-07
cal.ksamping = G
cal.kpoints = [3,3,3]
cal.sigma = 0.25
phonon.type = bandDos
phonon.structureSize = [2,2,2]
phonon.primitiveUVW = [0.0, 0.5, 0.5, 0.5, 0.0, 0.5, 0.5, 0.5, 0.0]
phonon.method = dfpt
phonon.qpoints = [41,41,41]
phonon.dosRange = [0,20]
phonon.qpointsLabel = [G,X,W,G,M]
phonon.qpointsCoord = [0.0, 0.0, 0.0, 0.5, 0.0, 0.0, 0.5, 0.5, 0.0, 0.0, 0.0, 0.0, 0.5, 0.5, 0.5]
phonon.qpointsNumber = 51
io.charge = false
io.wave = false
phonon.in 输入参数介绍:
在声子计算中可以尽量保留sys.和cal.的参数到 phonon.in 中,之后设置声子计算特有的参数即可:
task: 本次计算为声子计算,设置task为phonon;phonon.type: 表示声子计算哪些性质,bandDos对应计算声子能带和态密度;phonon.structureSize: 声子计算时超胞的大小;phonon.primitiveUVW: 表示声子能带计算时原胞UVW的系数;phonon.method: 表示声子计算的方式,dfpt为密度泛函微扰理论方法;phonon.qpoints: 表示声子计算q空间网格取样为41*41*41;phonon.dosRange: 表示声子态密度计算的能量区间为[0,20];phonon.qpointsLabel: 表示声子能带计算时高对称点标签;phonon.qpointsCoord: 表示声子能带计算时高对称点坐标;phonon.qpointsNumber: 表示声子能带相邻两个高对称点的间隔;
structure.as 文件参考如下:
Total number of atoms
8
Lattice
4.2555564654942897 0.0000000000000000 0.0000000000000000
0.0000000000000000 4.2555564654942888 0.0000000000000000
0.0000000000000000 0.0000000000000000 4.2555564654942897
Direct
Mg 0.0000000000000000 0.0000000000000000 0.0000000000000000
Mg 0.0000000000000000 0.5000000000000000 0.5000000000000000
Mg 0.5000000000000000 0.0000000000000000 0.5000000000000000
Mg 0.5000000000000000 0.5000000000000000 0.0000000000000000
O 0.5000000000000000 0.5000000000000000 0.5000000000000000
O 0.5000000000000000 0.0000000000000000 0.0000000000000000
O 0.0000000000000000 0.5000000000000000 0.0000000000000000
O 0.0000000000000000 0.0000000000000000 0.5000000000000000
注解
声子计算时应提高自洽计算的收敛精度,建议设置在1.0e-7以上。
计算声子之前建议先作结构弛豫计算(relax.freedom = all),便于得到准确的声子谱图。
声子计算时若打开对称性,建议适当提高对称性判断的精度,参数sys.symmetryAccuracy可设置为1.0e-6或更小,助于得到准确的计算结果。
phonon.iniPhonon可指定路径读取声子计算(phonon.type = phonon)得到的phonon.json文件,从而直接进行能带与态密度的计算。
phonon.type控制计算声子的类型,phonon对应计算声子,band对应计算声子能带,dos对应计算声子态密度,bandDos对应同时计算声子能带和态密度。当phonon.type = band/dos/bandDos且phonon.iniPhonon未指定文件路径时,程序先自动完成phonon.type = phonon的声子计算,然后根据任务计算能带或态密度。
2.16.2. run程序运行¶
准备好输入文件之后,将 phonon.in 和 structure.as 文件上传到服务器上运行,按照结构弛豫中介绍的方法执行 DS-PAW phonon.in 。
2.16.3. analysis计算结果分析¶
根据上述的输入文件,计算完成之后将会得到 DS-PAW.log 、 phonon.json 、 dfpt.json 和 dfpt.as 这4个文件。
dfpt.as :声子计算时超胞的结构文件,计算声子时读取该文件信息。
dfpt.json :声子计算时的参数文件,该文件与 phonon.in 文件信息一致,计算声子时读取该文件信息。
phonon.json :声子计算完成之后的 json 数据文件;此时声子能带数据被保存在 phonon.json 中,具体的数据结构详见数据结构解析部分;
使用 Device Studio 可直接对 phonon.json 文件处理出图,其操作步骤为:Simulator-->DS-PAW-->Analysis Plot,选择 phonon.json 即可,可根据作图要求自定义设置面板参数,处理得到的声子能带和态密度图如下(a)、(b)所示:
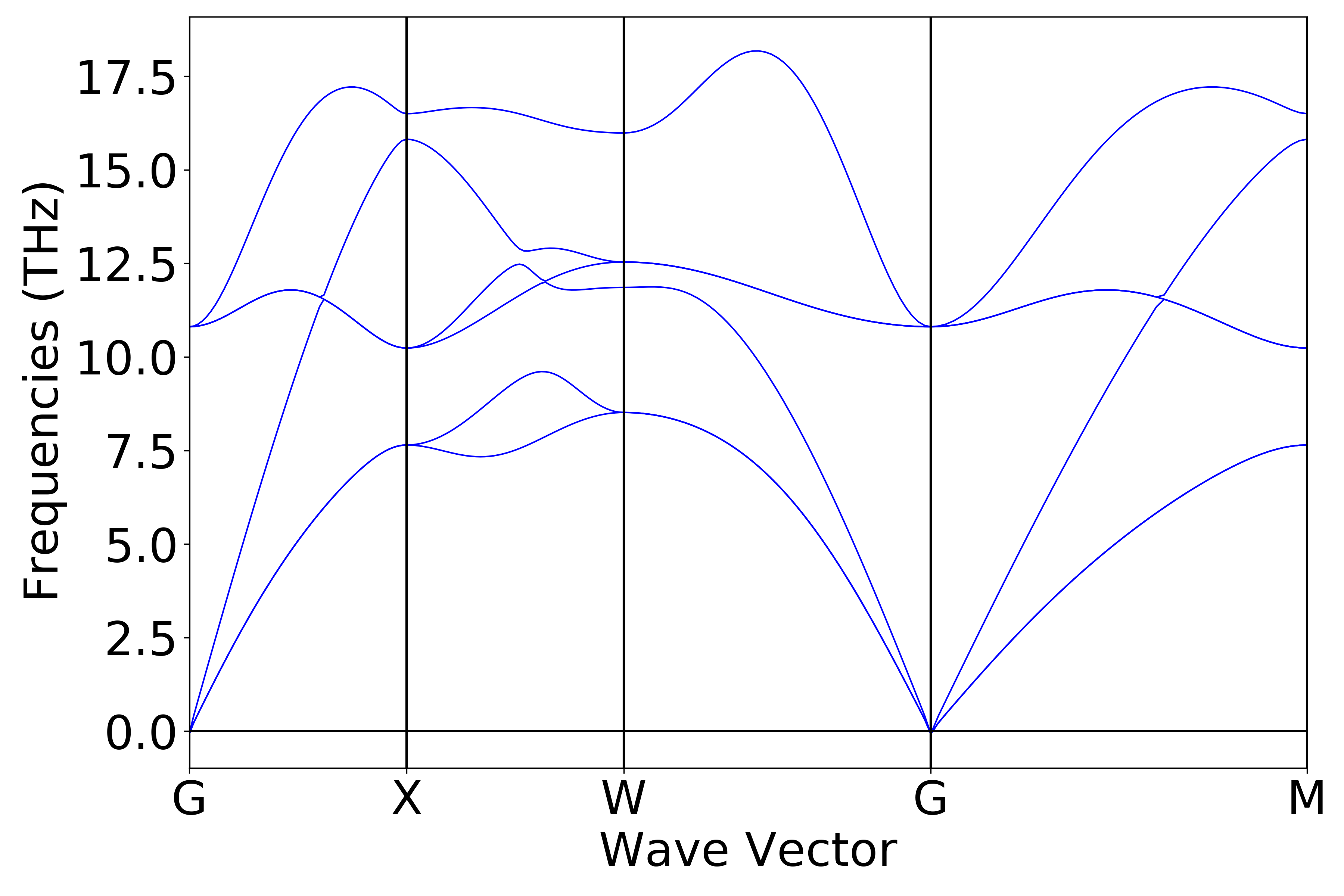
(a)
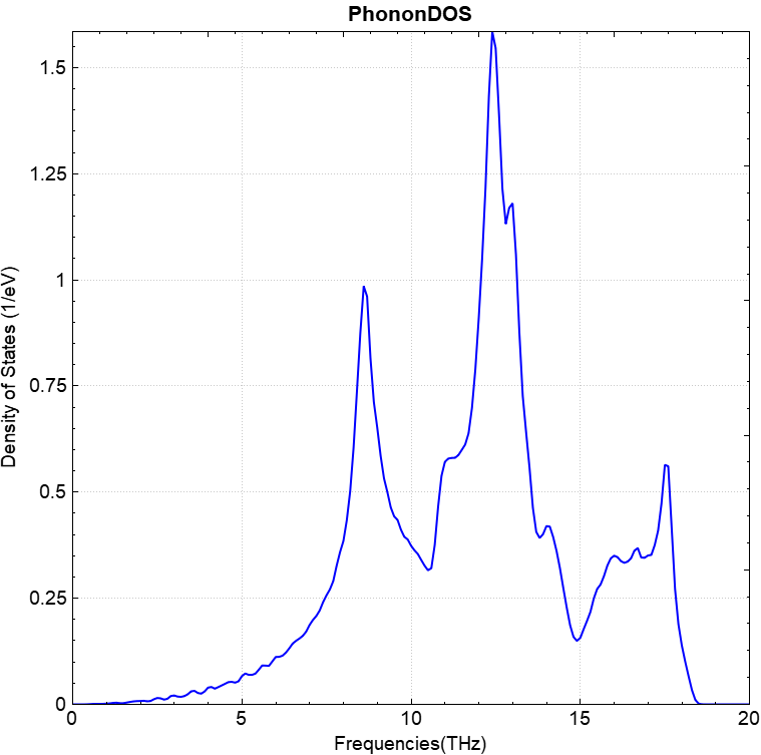
(b)
2.16.4. nac计算结果分析¶
上节展示的为不考虑长程相互作用的声子能带计算,若打开 non-analytical term correction (nac) 进行声子计算,在上节所示的 phonon.in 文件中添加以下两个参数即可:
phonon.dfptEpsilon=true
phonon.nac = true
得到的声子能带图如下(c)所示:
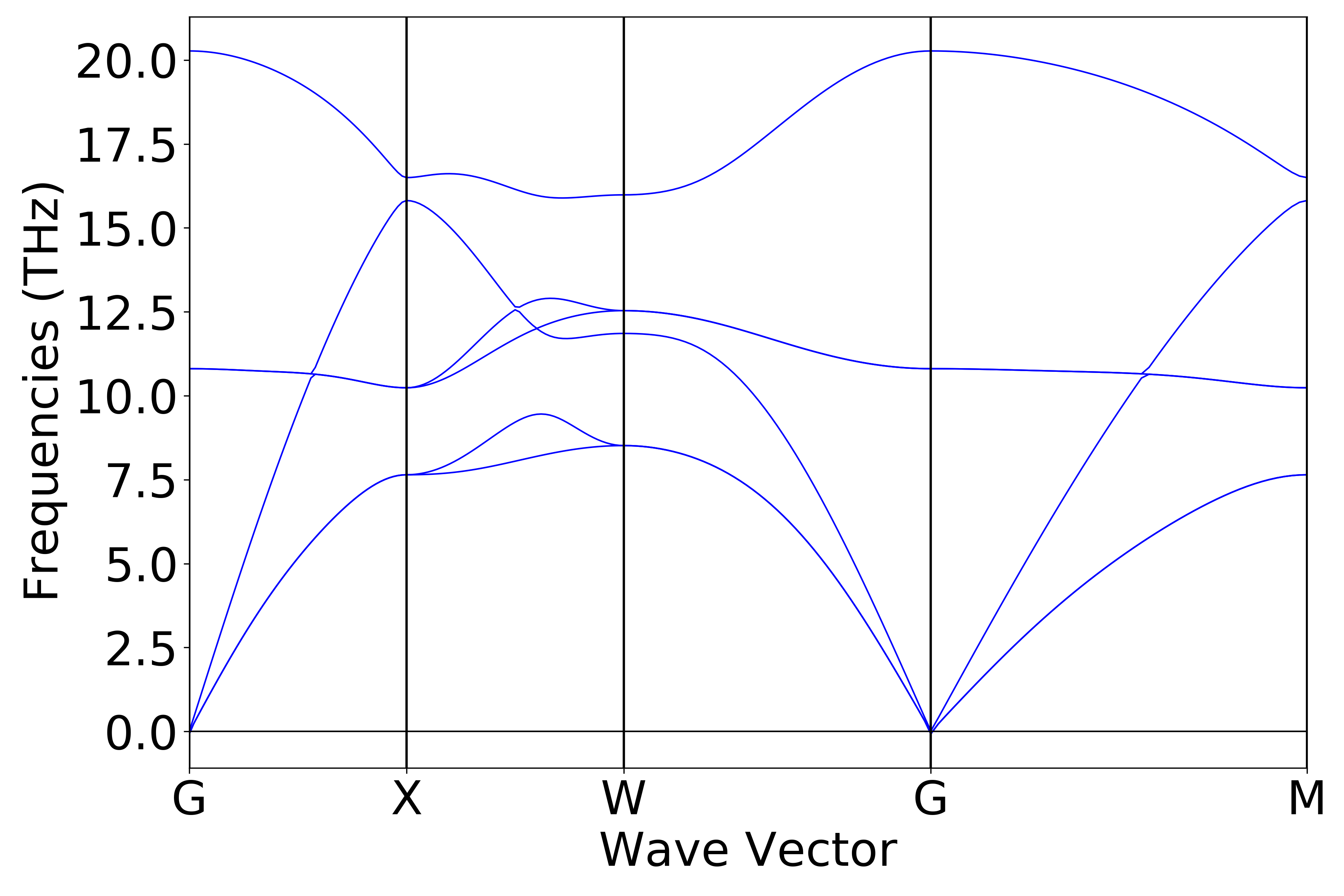
(c)
另可使用 python 进行数据处理,具体操作见 辅助工具使用教程 部分。
2.16.5. fdphonon有限位移法计算声子¶
有限位移法(fd) 计算的输入文件如下所示,将参数 phonon.method = dfpt 修改为 phonon.method = fd 即可,需要注意的是,fd法计算得到输出文件与dfpt法不同。
task = phonon
sys.structure = structure.as
sys.functional = PBE
sys.spin = none
cal.methods = 1
cal.smearing = 1
sys.symmetry = true
scf.convergence = 1.0e-07
cal.ksamping = G
cal.kpoints = [3,3,3]
cal.sigma = 0.25
phonon.type = bandDos
phonon.structureSize = [2,2,2]
phonon.primitiveUVW = [0.0, 0.5, 0.5, 0.5, 0.0, 0.5, 0.5, 0.5, 0.0]
phonon.method = fd
phonon.qpointsLabel = [G,X,W,G,M]
phonon.qpointsCoord = [0.0, 0.0, 0.0, 0.5, 0.0, 0.0, 0.5, 0.5, 0.0, 0.0, 0.0, 0.0, 0.5, 0.5, 0.5]
phonon.qpointsNumber = 51
io.charge = false
io.wave = false
以MgO体系为例, phonon.structureSize 设置为 [2,2,2] ,fd法计算完成之后将会得到 DS-PAW.log 、 phonon.json 两个文件 和 001 、 002 文件夹。 001 文件夹下存在 input.json 和 disp-001.as 文件,002 文件夹下存在 input.json 和 disp-002.as 文件,子文件夹中的两个文件等同于写入输入参数的 in 文件和结构参数的 as 文件,
生成文件夹(001 002...)的个数取决于体系的对称性。
用 Device Studio 处理fd法计算得到的 phonon.json 文件,得到能带图及态密度图同dfpt方法计算得到的图(a)与(b)一致。
注解
介电常数的计算只在phonon.method = dfpt时才能完成
phonon.nac的开关只在phonon.method = dfpt且phonon.dfptEpsilon=true时生效
2.17. soc自旋轨道耦合计算¶
本节介绍DS-PAW如何进行自旋轨道耦合计算。以 \(Bi_{2}Se_{3}\) 体系为例,使用两步法进行能带计算并对能带进行作图分析。
2.17.1. \(Bi_{2}Se_{3}\) 自旋轨道耦合计算输入文件¶
首先进行自洽计算:输入文件包含参数文件 soi.in 和结构文件 structure.as , soi.in 如下:
# task type
task = scf
#system related
sys.structure = structure.as
sys.symmetry = true
sys.functional = LDA
#scf related
cal.methods = 2
cal.smearing = 1
cal.ksamping = G
cal.kpoints = [7, 7, 7]
cal.cutoffFactor = 1.5
#soi related
sys.spin= non-collinear
sys.soi = true
#outputs
io.charge = true
io.wave = false
soi.in 输入参数介绍:
在自旋轨道耦合计算中可以尽量保留sys.和cal.的参数到 soi.in 中,之后设置自旋轨道耦合计算特有的参数即可:
soi.spin: 指定计算的自旋性质,non-collinear 表示一般自旋;sys.soi: 表示是否考虑自旋轨道耦合效应;自旋轨道耦合效应需要在sys.spin=non-collinear时才会生效;
structure.as 文件参考如下:
Total number of atoms
5
Lattice
-2.069 -3.583614 0.000000
2.069 -3.583614 0.000000
0.000 2.389075 9.546667
Direct
Bi 0.3990 0.3990 0.6970
Bi 0.6010 0.6010 0.3030
Se 0.0000 0.0000 0.5000
Se 0.2060 0.2060 0.1180
Se 0.7940 0.7940 0.8820
然后进行能带计算:输入文件包含参数文件 soiband.in ,内容如下
# task type
task = band
#system related
sys.structure = structure.as
sys.symmetry = true
sys.functional = LDA
#scf related
cal.methods = 2
cal.smearing = 1
cal.ksamping = G
cal.kpoints = [7, 7, 7]
cal.cutoffFactor = 1.5
#band related
cal.iniCharge = ./rho.bin
band.kpointsCoord = [0.00000000,0.00000000,0.00000000,0.00000000,0.00000000,0.50000000,0.50000000,0.50000000,0.00000000,0.00000000,0.00000000,0.00000000,0.50000000,0.00000000,0.00000000]
band.kpointsLabel = [G,Z,F,G,L]
band.kpointsNumber = [20,20,20,20]
band.project = true
#soi related
sys.spin= non-collinear
sys.soi = true
soiband.in 输入参数介绍:
在自旋轨道耦合能带计算中,保留自洽计算和自旋轨道耦合计算的参数到 soiband.in 中,之后设置能带计算的特有参数即可。
注解
初始磁矩的设置参考“应用案例-NiO体系的反铁磁计算”,在structure.as文件的第七行设置Mag标签即可。
2.17.2. run程序运行¶
准备好输入文件之后,将 soi.in 、 soiband.in 和 structure.as 文件上传到服务器上运行,按照结构弛豫中介绍的方法分别执行 DS-PAW soi.in 和 DS-PAW soiband.in 。
2.17.3. analysis计算结果分析¶
根据上述的输入文件,计算完成之后将会得到 DS-PAW.log 、 system.json 和 band.json 这3个文件。
处理 band.json 的方法同(见2.3节)能带计算的方法,DS处理得到的 bandplot.png 文件如下图(a)所示,另作不考虑自旋轨道耦合的计算,得到的能带图如下(b)所示:
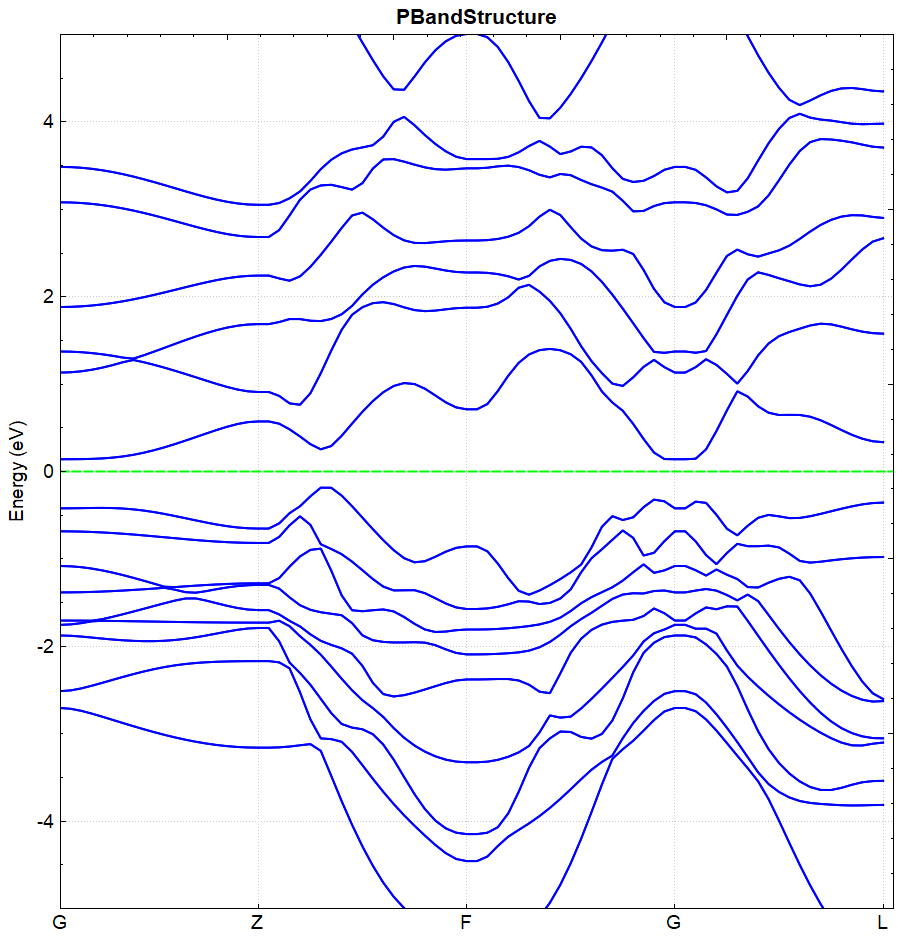
(a)
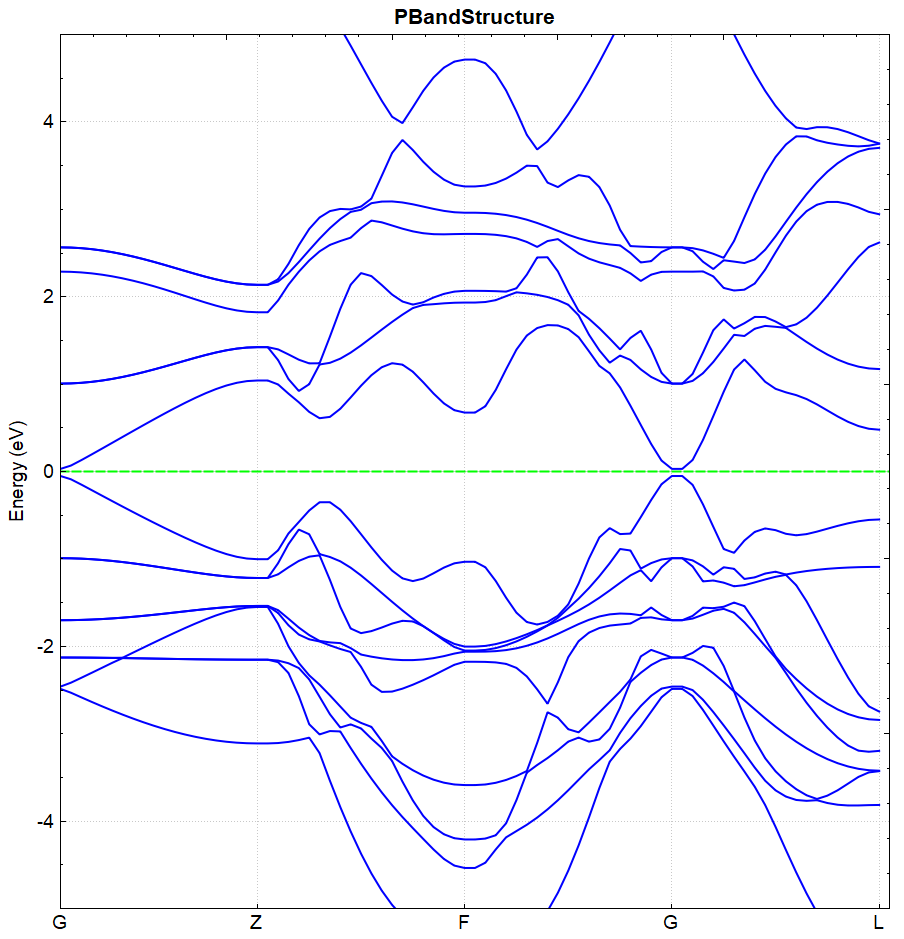
(b)
从 band.json 可读取 BandGap 值,图(a)和图(b)的带隙值分别为 0.3251 和 0.0814 ,可得结论:进行自旋轨道耦合计算可加大价带与导带之间的带隙。
2.18. aimd分子动力学模拟¶
本节将以水分子体系为例,介绍在DS-PAW中如何进行分子动力学模拟计算。
2.18.1. \(H_{2}O\) 分子动力学模拟输入文件¶
输入文件包含参数文件 aimd.in 和结构文件 structure.as , aimd.in 如下:
# task type
task = aimd
#system related
sys.structure = structure.as
sys.symmetry = false
sys.functional = LDA
sys.spin = none
#scf related
cal.methods = 1
cal.smearing = 2
cal.ksamping = G
cal.kpoints = [1, 1, 1]
cal.cutoffFactor = 1.5
#aimd related
aimd.ensemble = NVE
aimd.timeStep = 1
aimd.totalSteps = 1000
aimd.iniTemp = 2000
#outputs
io.charge = false
io.wave = false
aimd.in 输入参数介绍:
在分子动力学模拟计算中可以尽量保留sys.和cal.的参数到 aimd.in 中,之后设置分子动力学模拟计算特有的参数即可:
task: 本次计算为分子动力学计算,设置task为aimd;aimd.ensemble: 表示分子动力学模拟时选用的系综;aimd.timeStep: 表示分子动力学模拟时的时间步长;aimd.totalSteps: 表示分子动力学模拟的总步数;aimd.iniTemp: 表示分子动力学模拟时的初始温度;
structure.as 文件参考如下:
Total number of atoms
3
Lattice
6.35016 0 0
0 6.35016 0
0 0 6.35016
Direct
O 0.00000 0.00000 0.00000
H 0.09167 -0.11917 0.00000
H 0.09167 0.11917 0.00000
2.18.2. run程序运行¶
准备好输入文件之后,将 aimd.in 和 structure.as 文件上传到服务器上运行,按照结构弛豫中介绍的方法执行 DS-PAW aimd.in 。
2.18.3. analysis计算结果分析¶
根据上述的输入文件,计算完成之后将会得到 DS-PAW.log 、 aimd.json 、 paw_tmp/aimd.tmp 这3个文件。
aimd.json : 分子动力学计算完成之后的 json 数据文件;此时模拟时间内原子位置、体系能量和温度等数据被保存在 aimd.json 中,具体的数据结构详见数据结构解析部分;
paw_tmp/aimd.tmp : 分子动力学计算中的轨迹文件,默认aimd计算中每20个离子步记录一次结构信息;
使用 Device Studio 可直接对 aimd.json 文件处理出图,其操作步骤为:Simulator-->DS-PAW-->Analysis Plot,选择 aimd.json 即可,可根据作图要求自定义设置面板参数。DS可以播放在模拟过程中分子的运动轨迹,截取其中一帧的结构图如下图所示:

另可使用 python 进行数据处理,将 aimd.json 转为 pdb 文件,具体操作见 辅助工具使用教程 部分。
注解
本例设置的系综为nve,设置nvt系综时可选择程序支持的两种控温器,通过 aimd.thermostat 参数设置;
如需模拟高温退火过程,设置 aimd.ensemble 为 TS,同时通过 aimd.iniTemp 和 aimd.iniTemp 设置初始和末态温度即可。
2.19. efield外加电场计算¶
本节将以硅烯模型为例,介绍在DS-PAW中如何进行外加电场计算,分析加电场前后带隙打开情况。
2.19.1. Si硅烯真空方向外加电场计算输入文件¶
输入文件包含参数文件 Efield.in 和结构文件 structure.as , Efield.in 如下:
# task type
task = scf
#system related
sys.structure = structure.as
sys.symmetry = true
sys.functional = PBE
sys.spin = none
#scf related
cal.sigma = 0.1
cal.cutoff = 520
cal.ksamping = G
cal.kpoints = [9, 9, 1]
scf.convergence = 1e-5
#outputs
io.charge = false
io.wave = false
io.band = true
corr.dipol=true
corr.dipolDirection = c
corr.dipolEfield = 0.2
band.kpointsLabel = [G,M,K,G]
band.kpointsCoord = [0.00000000,0.00000000,0.00000000,0.50000000,0.00000000,0.00000000,0.33333333,0.33333333,0.00000000,0.00000000,0.00000000,0.00000000]
band.kpointsNumber = [100,100,100]
Efield.in 输入参数介绍:
该计算是在一步能带计算的基础上外加电场,除能带计算的基本参数,新增参数为下:
corr.dipolEfield: 设置外加电场的大小,注意该参数只在corr.dipol = true和设置corr.dipolDirection的情况下生效;
structure.as 文件参考如下:
Total number of atoms
2
Lattice
3.860000 0.000000 0.000000
-1.930000 3.342860 0.000000
0.000000 0.000000 26.460000
Direct
Si 0.333333 0.166667 0.396825
Si 0.666758 0.833380 0.379216
2.19.2. run程序运行¶
准备好输入文件之后,将 Efield.in 和 structure.as 文件上传到服务器上运行,按照结构弛豫中介绍的方法执行 DS-PAW Efield.in 。
2.19.3. analysis计算结果分析¶
根据上述的输入文件,计算完成之后将会得到 DS-PAW.log 、 band.json 这2个文件。
band.json :计算完成之后的 json 数据文件;
此例中参数 corr.dipolEfield = 0.2 ,即外加电场的大小为 0.2 eV/Å, 在该电场下进行能带计算得到的能带图如图(a)所示,
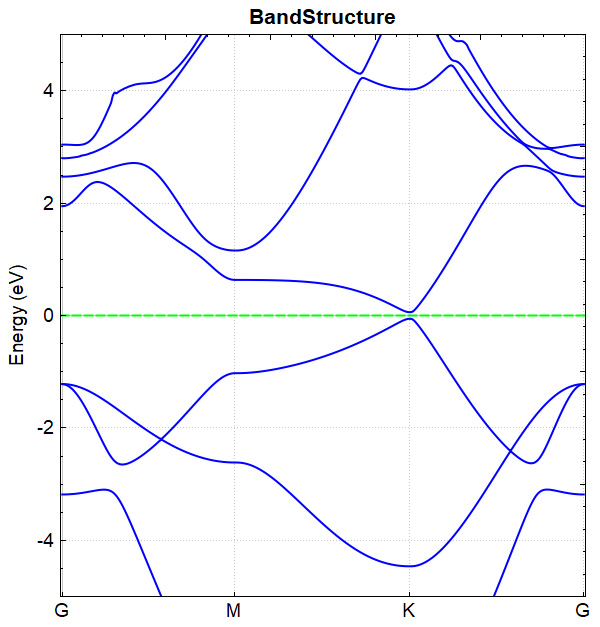
(a)
若设置参数 corr.dipolEfield = 0 重复以上计算,即在无电场的情况下进行能带计算得到的能带图如图(b)所示,
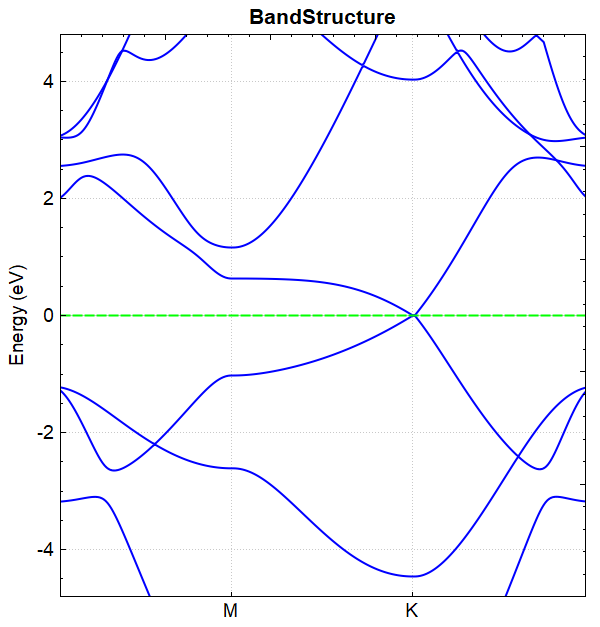
(b)
对比图(a)和图(b)可得结论:通过外加电场可以打开硅烯的带隙。 从 band.json 文件可读出加电场与不加电场 BandGap 的值分别为 0.1176 和 0.0010 。
注解
外加电场的单位 eV/Å 为原子受力的单位
2.20. ferri铁电计算¶
本节将以 \(HfO_{2}\) 为例,介绍在DS-PAW中如何使用现代极化理论进行铁电计算,分析 \(HfO_{2}\) 的铁电极化。
2.20.1. \(HfO_{2}\) 铁电计算输入文件¶
输入文件包含参数文件 polarization.in 和结构文件 structure.as , polarization.in 如下:
# task type
task = scf
#system related
sys.structure = structure.as
sys.symmetry = true
sys.functional = PBE
sys.spin = none
#scf related
cal.methods = 3
cal.smearing = 4
cal.sigma = 0.05
cal.cutoff = 520
cal.ksamping = MP
cal.kpoints = [4, 4, 4]
scf.convergence = 1e-5
#outputs
io.charge = false
io.wave = false
io.polarization = true
polarization.in 输入参数介绍:
该计算是在自洽计算的基础上进行铁电计算,除自洽计算的基本参数,新增参数为下:
io.polarization: 在自洽计算中打开控制铁电计算的开关,即可快速实现铁电计算;
structure.as 文件参考如下:
Total number of atoms
12
Lattice
5.04621935 0.00000000 0.00000000
0.00000000 5.07315250 0.00000000
0.00000000 0.00000000 5.25768906
Cartesian
Hf 1.34815269 1.22145223 0.17639072
Hf 1.34815269 3.75802848 2.45245381
Hf 3.69806666 1.22145223 2.80523525
Hf 3.69806666 3.75802848 5.08129834
O 0.35195212 1.93667285 1.92589951
O 0.35195212 4.47324910 0.70294502
O 2.32678304 2.48829365 3.85528784
O 2.32678304 5.02486990 4.03124575
O 2.71943630 5.02486990 1.40240122
O 2.71943630 2.48829365 1.22644331
O 4.69426723 1.93667285 4.55474404
O 4.69426723 4.47324910 3.33178955
2.20.2. run程序运行¶
准备好输入文件之后,将 polarization.in 和 structure.as 文件上传到服务器上运行,按照结构弛豫中介绍的方法执行 DS-PAW polarization.in 。
2.20.3. analysis计算结果分析¶
根据上述的输入文件,计算完成之后将会得到 DS-PAW.log 、 polarization.json 和 polarization.txt 这3个文件。
polarization.json :铁电极化计算完成之后的 json 数据文件,电子、离子贡献的极化部分及总的极化量子数存储在该文件中,具体文件解析见输出文件格式说明部分;
polarization.txt : 铁电极化计算完成之后的 txt 文本文件,该文件写入极化数据,与 polarization.json 文件数据一致,便于用户快速获取信息。
从 polarization.txt 文件可得 \(HfO_{2}\) 的铁电极化数据如下所示:
Total(x y z) ((μC/cm^2))
-0.000001
-8.715113
0.000000
Quantum(x y z) (μC/cm^2)
60.067225
60.387821
62.584436
通过极化量子的周期性换算,可得y方向上 \(HfO_{2}\) 的极化数为 51.672 μC/cm^2 ,与文献报道结果[2] 53 μC/cm^2 一致。
2.21. bader电荷计算¶
本节将以NaCl晶体为例,介绍在DS-PAW中如何进行bader电荷计算,分析NaCl体系中各原子的价态分布。
2.21.1. \(NaCl\) 晶体Bader电荷计算输入文件¶
输入文件包含参数文件 bader.in 和结构文件 structure.as , bader.in 如下:
bader.in 文件参考如下:
# task type
task = scf
#system related
sys.structure = structure.as
sys.symmetry = false
sys.functional = PBE
sys.spin = none
#scf related
cal.methods = 1
cal.smearing = 1
cal.ksamping = G
cal.kpoints = [10, 10, 10]
cal.cutoff = 500
#outputs
io.charge = true
io.wave = false
io.bader = true
bader.in 输入参数介绍:
该计算是在自洽计算的基础上进行bader电荷计算,除自洽计算的基本参数,新增参数为下:
io.bader: 在自洽计算中打开控制bader电荷计算的开关,即可快速实现bader电荷计算;
structure.as 文件参考如下:
Total number of atoms
8
Lattice
5.68452692 0.00000000 0.00000000
0.00000000 5.68452692 0.00000000
0.00000000 0.00000000 5.68452692
Cartesian
Na 4.26339519 1.42113173 1.42113173
Na 1.42113173 4.26339519 1.42113173
Na 1.42113173 1.42113173 4.26339519
Na 4.26339519 4.26339519 4.26339519
Cl 1.42113173 1.42113173 1.42113173
Cl 4.26339519 4.26339519 1.42113173
Cl 4.26339519 1.42113173 4.26339519
Cl 1.42113173 4.26339519 4.26339519
注解
io.bader = true 时,io.charge 必须设置为true
2.21.2. run程序运行¶
准备好输入文件之后,将 bader.in 和 structure.as 文件上传到服务器上运行,按照结构弛豫中介绍的方法执行 DS-PAW bader.in 。
2.21.3. analysis计算结果分析¶
根据上述的输入文件,计算完成之后将会得到 DS-PAW.log 、 bader.json 、 bader.txt 这3个文件。
bader.json :bader电荷计算完成之后的 json 数据文件,各原子的Bader电荷等数据存储在该文件中,具体的数据结构详见 输出文件格式说明 部分;
bader.txt : bader电荷计算完成之后的 txt 文本文件,该文件写入bader电荷数据,与 bader.json 文件数据一致,便于用户快速获取信息。
bader.txt 文本内容如下所示,bader电荷分析得到的数据与utexas大学的Henkelman小组得到的数据吻合。https://theory.cm.utexas.edu/henkelman/code/bader/
Total number of valence electronics: 64
Element |
X |
Y |
Z |
Charge |
AtomicVolume |
MinDistance |
Cl |
0.25 |
0.25 |
0.25 |
7.83165 |
34.846 |
1.59877 |
Cl |
0.75 |
0.75 |
0.25 |
7.87519 |
36.8613 |
1.59877 |
Cl |
0.75 |
0.25 |
0.75 |
7.84614 |
35.5159 |
1.59877 |
Cl |
0.25 |
0.75 |
0.75 |
7.86064 |
36.1858 |
1.59877 |
Na |
0.75 |
0.25 |
0.25 |
8.1466 |
10.07 |
1.11292 |
Na |
0.25 |
0.75 |
0.25 |
8.1466 |
10.07 |
1.11292 |
Na |
0.25 |
0.25 |
0.75 |
8.1466 |
10.07 |
1.11292 |
Na |
0.75 |
0.75 |
0.75 |
8.1466 |
10.07 |
1.11292 |
2.22. bandunfolding能带反折叠计算¶
本节将以 \(Cu_{3}Au\) 体系为例,介绍在DS-PAW中如何进行能带反折叠计算,分析 \(Cu_{3}Au\) 反折叠的能带图。
2.22.1. \(Cu_{3}Au\) 能带反折叠计算输入文件¶
能带反折叠计算需两步法完成能带计算,因此输入文件包含参数文件 scf.in 、 bandunfolding.in 和结构文件 structure.as ,
scf.in 如下:
task = scf
sys.structure = structure.as
sys.symmetry = true
sys.functional = PBE
sys.spin = none
cal.methods = 1
cal.smearing = 1
cal.ksamping = MP
cal.kpoints = [3, 3, 3]
cal.cutoff = 500
scf.convergence = 1.0e-05
io.charge = true
io.wave = false
bandunfolding.in 如下:
task = band
cal.iniCharge = ./rho.bin
sys.structure = structure.as
sys.symmetry = true
sys.functional = PBE
sys.spin = none
cal.methods = 1
cal.smearing = 1
cal.ksamping = MP
cal.kpoints = [3, 3, 3]
cal.cutoff = 500
scf.convergence = 1.0e-05
band.unfolding = true
band.primitiveUVW=[0.0, 0.5, 0.5, 0.5, 0.0, 0.5, 0.5, 0.5, 0.0]
band.kpointsLabel= [R,G,X]
band.kpointsCoord= [0.5, 0.5, 0.5, 0.0, 0.0, 0.0, 0.5, 0.0, 0.5]
band.kpointsNumber= [101, 101]
io.charge = false
io.wave = false
bandunfolding.in 输入参数介绍:
能带反折叠计算是在能带计算的基础上完成的,且能带计算必须通过两步法完成。除能带计算的基本参数,新增参数为下:
band.unfolding: 在能带计算中打开能带反折叠计算的开关,即可实现能反折叠计算;band.primitiveUVW: 超胞的晶格常数乘上UVW系数等于原胞的晶格矢量,用于控制能带反折叠的参数;
structure.as 文件参考如下:
Total number of atoms
4
Lattice
3.7530000210 0.0000000000 0.0000000000
0.0000000000 3.7530000210 0.0000000000
0.0000000000 0.0000000000 3.7530000210
Direct
Au 0.000000000 0.000000000 0.000000000
Cu 0.000000000 0.500000000 0.500000000
Cu 0.500000000 0.000000000 0.500000000
Cu 0.500000000 0.500000000 0.000000000
2.22.2. run程序运行¶
准备好输入文件之后,将 scf.in 、bandunfolding.in 和 structure.as 文件上传到服务器上运行,按照结构弛豫中介绍的方法执行 DS-PAW scf.in ,自洽计算完成后执行 DS-PAW bandunfolding.in 。
2.22.3. analysis计算结果分析¶
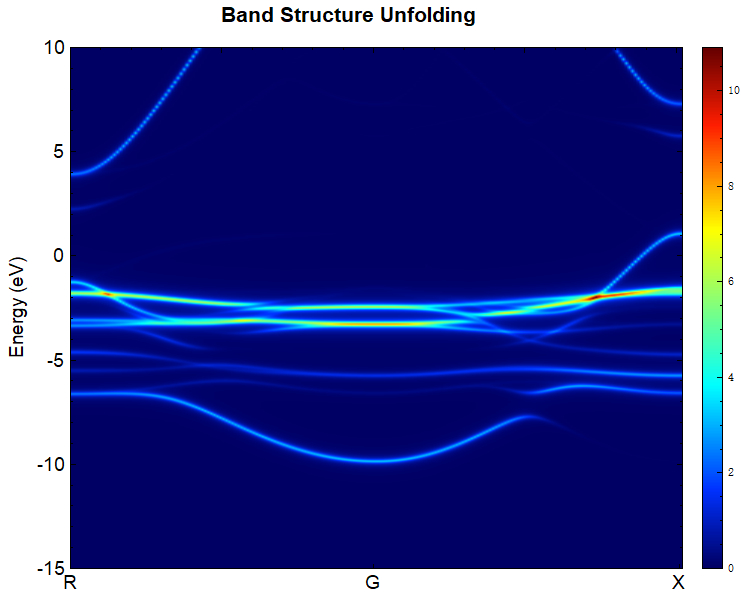
根据上述的输入文件,计算完成之后将会得到 DS-PAW.log 、 band.json 、 log 这3个文件。
band.json :计算完成之后的 json 数据文件,能带反折叠计算所得json文件与普通能带计算不同,会新增 UnfoldingBandInfo 部分,具体结构解析见输出文件格式说明部分。
使用 Device Studio 可直接对 band.json 文件处理出图,其操作步骤为:Simulator-->DS-PAW-->Analysis Plot,选择 band.json 即可,可根据作图要求自定义设置面板参数。
该例得到的能带图如下所示,与文献报道结果[3] 一致。
2.23. epsilon介电常数计算¶
本节将以Si体系为例,介绍在DS-PAW中如何进行介电常数计算。
2.23.1. \(Si\) 介电常数计算输入文件¶
输入文件包含参数文件 epsilon.in 和结构文件 structure.as , epsilon.in 如下:
# task type
task = epsilon
#system related
sys.structure = structure.as
sys.symmetry = true
sys.functional = PBE
sys.spin = none
#scf related
cal.methods = 1
cal.smearing = 1
cal.ksamping = G
cal.kpoints = [5, 5, 5]
cal.cutoff = 500
scf.convergence = 1.0e-7
epsilon.in 输入参数介绍:
介电常数的计算可通过直接指定 task 完成,新增参数为下:
task = epsilon: 通过控制任务类型完成介电常数的计算;
注解
在 task = phonon 且 phonon.method = dfpt 时也可完成介电常数的计算,通过添加 phonon.dfptEpsilon = true 参数即可。
structure.as 文件参考如下:
Total number of atoms
8
Lattice
5.43070000 0.00000000 0.00000000
0.00000000 5.43070000 0.00000000
0.00000000 0.00000000 5.43070000
Cartesian
Si 0.67883750 0.67883750 0.67883750
Si 3.39418750 3.39418750 0.67883750
Si 3.39418750 0.67883750 3.39418750
Si 0.67883750 3.39418750 3.39418750
Si 2.03651250 2.03651250 2.03651250
Si 4.75186250 4.75186250 2.03651250
Si 4.75186250 2.03651250 4.75186250
Si 2.03651250 4.75186250 4.75186250
2.23.2. run程序运行¶
准备好输入文件之后,将 epsilon.in 和 structure.as 文件上传到服务器上运行,按照结构弛豫中介绍的方法执行 DS-PAW epsilon.in 。
2.23.3. analysis计算结果分析¶
根据上述的输入文件,计算完成之后将会得到 DS-PAW.log 、 epsilon.json 、 epsilon.txt 这3个文件。
epsilon.json :介电常数计算完成之后的 json 数据文件,介电常数、波恩有效电荷等数据会写入该文件,具体的数据结构详见 输出文件格式说明 部分;
epsilon.txt : 介电常数计算完成之后的 txt 文本文件,该文件写入介电常数相关数据,与 epsilon.json 文件数据一致,便于用户快速获取信息。
从 epsilon.txt 文件中可获取如下数据:
Total Part |
||
13.309902 |
0.000000 |
-0.000000 |
-0.000000 |
13.309902 |
-0.000000 |
-0.000000 |
0.000000 |
13.309902 |
分析上表可得该体系的介电常数为 13.309902 ,与文献报道值[4] 13.31 一致。
2.24. piezo压电张量计算¶
本节将以AlN体系为例,介绍在DS-PAW中如何进行压电张量计算,得到材料的压电系数 \(e_{33} (0)\)。
2.24.1. \(AlN\) 压电张量计算输入文件¶
输入文件包含参数文件 piezo.in 和结构文件 structure.as , piezo.in 如下:
task = epsilon
#system related
sys.structure = structure.as
sys.symmetry = true
sys.functional = PBE
sys.spin = none
#scf related
cal.methods = 1
cal.smearing = 1
cal.ksamping = G
cal.kpoints = [10, 10, 10]
cal.cutoffFactor = 1.5
scf.convergence = 1.0e-7
#outputs
io.charge = false
io.wave = false
piezo.in 输入参数介绍:
task: 本次计算为压电张量计算,设置task为epsilon;scf.convergence: 介电张量计算建议提高电子收敛的精度,此处设置为1.0e-7;
structure.as 文件参考如下:
Total number of atoms
8
Lattice
3.11606630 0.00000000 0.00000000
0.00000000 5.39683518 0.00000000
0.00000000 0.00000000 5.00770902
Cartesian
Al 0.00000000 3.59735137 0.00946380
Al 0.00000000 1.79945276 2.51320124
Al 1.55803315 0.89899597 0.00945662
Al 1.55803315 4.49786165 2.51308138
N 0.00000000 3.59851112 1.91845914
N 0.00000000 1.79831356 4.42266820
N 1.55803315 0.90013952 1.91851680
N 1.55803315 4.49672497 4.42258192
2.24.2. run程序运行¶
准备好输入文件之后,将 piezo.in 和 structure.as 文件上传到服务器上运行,按照结构弛豫中介绍的方法执行 DS-PAW piezo.in 。
2.24.3. analysis计算结果分析¶
根据上述的输入文件,计算完成之后将会得到 DS-PAW.log 、 epsilon.json 、 epsilon.txt 这3个文件。
epsilon.json :介电常量计算完成之后的 json 数据文件;介电常数、压电张量矩阵、波恩有效电荷的数据被保存在 epsilon.json 中,具体的数据结构详见数据结构解析部分;
epsilon.txt : 压电计算完成之后的 txt 文本文件,该文件写入压电相关数据,与 epsilon.json 文件数据一致,便于用户快速获取信息。
从 epsilon.txt 文件中可获取如下数据:
Piezoelectric Tensor (C/m^2)( Row: x y z Column: XX YY ZZ XY YZ ZX) |
|||||
Electronic Part |
|||||
0.000000 |
0.000000 |
0.000000 |
0.000006 |
0.000000 |
0.336610 |
-0.000001 |
0.000007 |
0.000003 |
0.000000 |
0.336662 |
0.000000 |
0.266339 |
0.265888 |
-0.419569 |
0.000000 |
-0.000014 |
0.000000 |
Ionic Part: |
|||||
-0.000004 |
0.000002 |
0.000002 |
0.000032 |
-0.000000 |
-0.681702 |
-0.000163 |
-0.000239 |
0.000314 |
-0.000000 |
-0.699012 |
-0.000000 |
-0.911456 |
-0.913265 |
1.943887 |
-0.000000 |
-0.000633 |
-0.000000 |
Total Part: |
|||||
-0.000004 |
0.000002 |
0.000002 |
0.000039 |
-0.000000 |
-0.345092 |
-0.000164 |
-0.000232 |
0.000317 |
-0.000000 |
-0.362350 |
-0.000000 |
-0.645117 |
-0.647377 |
1.524318 |
-0.000000 |
-0.000647 |
-0.000000 |
分析上表可得压电张量电子贡献部分 \(e_{33} (0)\) 的数值为 -0.419569 \(C/m^{2}\) ,总的压电张量 \(e_{33}\) 的数值为 1.524318 \(C/m^{2}\) ,与文献参考值[5] -0.47 \(C/m^{2}\) 和 1.46 \(C/m^{2}\) 接近。
2.25. fixcell固定基矢弛豫计算¶
本节将以 \(MoS_{2}\) 体系为例,介绍在DS-PAW中如何进行固定晶格弛豫计算。
2.25.1. \(MoS_{2}\) 固定基矢弛豫计算输入文件¶
输入文件包含参数文件 relax.in 和结构文件 structure.as , relax.in 如下:
# task type
task = relax
#system related
sys.structure = structure.as
sys.symmetry = false
sys.functional = PBE
sys.spin = none
#scf related
cal.methods = 1
cal.smearing = 1
cal.ksamping = G
cal.cutoff = 500
cal.kpoints = [19, 19, 5]
#relax related
relax.freedom = all
relax.convergence = 0.05
relax.methods = CG
structure.as 文件参考如下:
Total number of atoms
6
Lattice Fix_x Fix_y Fix_z
3.19031572 0.00000000 0.00000000 F T T
-1.59515786 2.76289446 0.00000000 F F T
0.00000000 0.00000000 14.87900448 T T T
Cartesian
S 0.00000000 1.84193052 12.72413785
S 1.59515943 0.92096386 5.28463561
S 0.00000000 1.84193052 9.59436887
S 1.59515943 0.92096386 2.15486663
Mo 1.59515943 0.92096386 11.15925336
Mo 0.00000000 1.84193052 3.71975112
structure.as 标签设置介绍:
固定晶胞维度进行弛豫计算需在 structure.as 文件中新增固定标签,类似于固定原子弛豫的设置(在原子坐标后添加Fix标签),固定基矢需在 structure.as 的第三行 Lattice 后添加Fix标签,本案例的标签对应固定晶胞的c边和a边的y、z方向、b边的z方向。
2.25.2. run程序运行¶
准备好输入文件之后,将 relax.in 和 structure.as 文件上传到服务器上运行,按照结构弛豫中介绍的方法执行 DS-PAW relax.in 。
2.25.3. analysis计算结果分析¶
根据上述的输入文件,计算完成之后将会得到 DS-PAW.log 、 relax.json 这2个文件。
relax.json :计算完成之后的 json 数据文件;
将 relax.json 拖入Device Studio查看结构,或直接查看 paw_tmp 目录下的relax.tmp 文件,可得弛豫结束后的结构数据如下:
Total number of atoms
6
Lattice
3.19696732 0.00000000 0.00000000
-1.59848077 2.76865753 0.00000000
0.00000000 0.00000000 14.87900448
Direct
Mo 0.66666701 0.33333316 0.74999995
Mo 0.33333340 0.66666675 0.24999997
S 0.33333340 0.66666666 0.85535854
S 0.66666686 0.33333303 0.35535875
S 0.33333367 0.66666699 0.64464148
S 0.66666708 0.33333333 0.14464130
通过对比可得弛豫前 a = b = 3.19031572 ,弛豫后变成了a = b = 3.19696732 ,而c = 14.87900448 不变。
2.26. thermal声子热力学性质计算¶
本节将以Si体系为例,介绍在DS-PAW中如何进行声子热力学性质计算。
2.26.1. \(Si\) 声子热力学性质计算输入文件¶
输入文件包含参数文件 phonon-thermal.in 和结构文件 structure.as , phonon-thermal.in 如下:
# task type
task = phonon
#system related
sys.structure = structure.as
sys.symmetry = true
sys.functional = PBE
sys.spin = none
#scf related
cal.methods = 1
cal.smearing = 1
cal.ksamping = G
cal.kpoints = [5, 5, 5]
cal.cutoffFactor = 1.5
scf.convergence = 1.0e-7
#phonon related
phonon.structureSize = [2,2,2]
phonon.type =dos
phonon.qpoints = [31,31,31]
phonon.method = dfpt
phonon.thermal=true
phonon.thermalRange = [0,1000,10]
phonon-thermal.in 输入参数介绍:
phonon.thermal: 该参数为控制热力学计算的开关;phonon.thermalRange: 该参数可指定热力学计算的温度范围及数据存储间隔;
structure.as 文件参考如下:
Total number of atoms
2
Lattice
0.00 2.75 2.75
2.75 0.00 2.75
2.75 2.75 0.00
Direct
Si -0.125000000 -0.125000000 -0.125000000
Si 0.125000000 0.125000000 0.125000000
2.26.2. run程序运行¶
准备好输入文件之后,将 phonon-thermal.in 和 structure.as 文件上传到服务器上运行,按照结构弛豫中介绍的方法执行 DS-PAW phonon-thermal.in 。
2.26.3. analysis计算结果分析¶
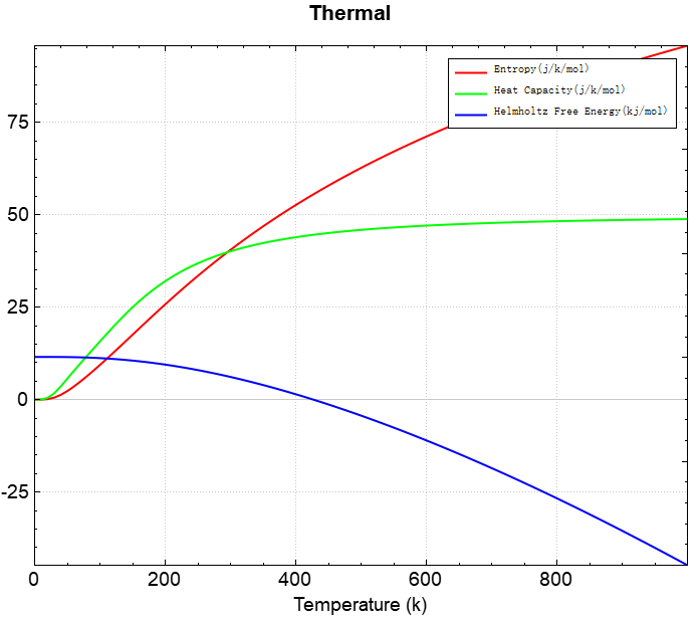
根据上述的输入文件,计算完成之后将会得到 DS-PAW.log 、 phonon.json 这2个文件。
phonon.json :计算完成之后生成的json文件,打开热力学计算的开关,生成的 phonon.json 文件中会写入 ThermalInfo 数据,具体解析见输出文件格式说明部分。
使用 Device Studio 可直接对 phonon.json 文件处理出图,其操作步骤为:Simulator-->DS-PAW-->Analysis Plot,选择 phonon.json 即可,可根据作图要求自定义设置面板参数。
分析热力学数据,可得熵、热容、亥姆霍兹自由能随温度变化的曲线如下所示,与phonony git仓库展示的结果一致:https://phonopy.github.io/phonopy/examples#thermal-properties
2.27. solid state NEB计算¶
本节将以HfZrO体系为例,介绍在DS-PAW中如何放开晶胞弛豫进行solid state NEB计算。
2.27.1. \(HfZrO\) Solid state NEB计算输入文件¶
输入文件包含参数文件 ssneb.in 和结构文件 structure.as , ssneb.in 如下:
task = neb
sys.structure = structure.as
sys.functional = LDA
sys.spin = none
sys.symmetry = false
cal.ksamping = G
cal.kpoints = [10,10,10]
cal.cutoff = 500
cal.methods = 1
cal.smearing = 1
cal.sigma = 0.05
scf.mixType = Broyden
scf.mixBeta = 0.4
scf.convergence = 1e-6
scf.max = 300
neb.springK = 5
neb.images = 6
neb.iniFin = true
neb.method = QM2
neb.convergence = 0.01
neb.max = 500
neb.freedom = all
io.wave = false
io.charge = false
ssneb.in 输入参数介绍:
neb.freedom: 该参数控制过渡态弛豫的维度,设置为all对应弛豫晶胞大小;neb.method: 该参数指定过渡态搜寻的方法,当弛豫晶胞大小时该参数仅限于QM2和FIRE;
structure.as 需提供多个,初态结构 structure00.as 参考如下
Total number of atoms
12
Lattice
5.00209138 0.00000009 0.00000004
0.00000009 5.00209143 -0.00000004
0.00000004 -0.00000004 5.07896990
Cartesian
Hf 2.50104558 2.50104575 0.00000000
Hf 0.00000000 0.00000000 0.00000000
O 3.75156841 1.25052303 1.47285183
O 3.75156857 3.75156869 1.04735062
O 1.25052293 1.25052297 3.60611823
O 1.25052286 3.75156867 4.03161932
O 1.25052287 3.75156860 1.47285187
O 1.25052275 1.25052294 1.04735054
O 3.75156850 1.25052287 4.03161945
O 3.75156850 3.75156869 3.60611821
Zr 2.50104577 0.00000000 2.53948497
Zr 0.00000000 2.50104594 2.53948491
末态结构 structure07.as 参考如下
Total number of atoms
12
Lattice
4.98221520 -0.00002552 0.00036684
-0.00002562 4.99587652 0.00005905
0.00039053 0.00006126 5.18258321
Cartesian
Hf 2.30823006 2.49975412 0.04967381
Hf 0.00919001 0.00195723 0.38722458
O 4.03365086 0.66419181 2.12958714
O 4.00001549 3.18954023 0.89210846
O 0.95871628 1.24120307 4.04442128
O 0.94984693 3.74053908 4.19050825
O 1.35895285 3.73907584 1.57483409
O 1.36804279 1.24264997 1.42944278
O 3.29999107 0.69159253 4.72728663
O 3.26626721 3.16200890 3.48972595
Zr 2.31915914 0.00841995 2.97686955
Zr 4.98082249 2.50639160 2.64290889
初末态构型在 Device Studio 中显示如下:
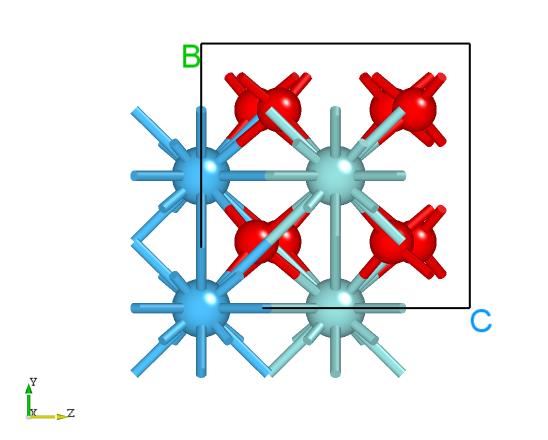
初态T相构型
末态F相构型
注解
在 neb.freedom = all 时, neb.method 只能设置为 QM2 或 FIRE
2.27.2. run程序运行¶
准备好输入文件之后,将 ssneb.in 和 structure.as 文件上传到服务器上运行,按照结构弛豫中介绍的方法执行 DS-PAW ssneb.in 。
2.27.3. analysis计算结果分析¶
根据上述的输入文件,计算完成之后将会得到 DS-PAW.log 、 neb.json 这2个文件。
neb.json :过渡态计算完成之后的 json 数据文件;此时反应坐标及能量变化等数据被保存在 neb.json 中,具体的数据结构详见数据结构解析部分;
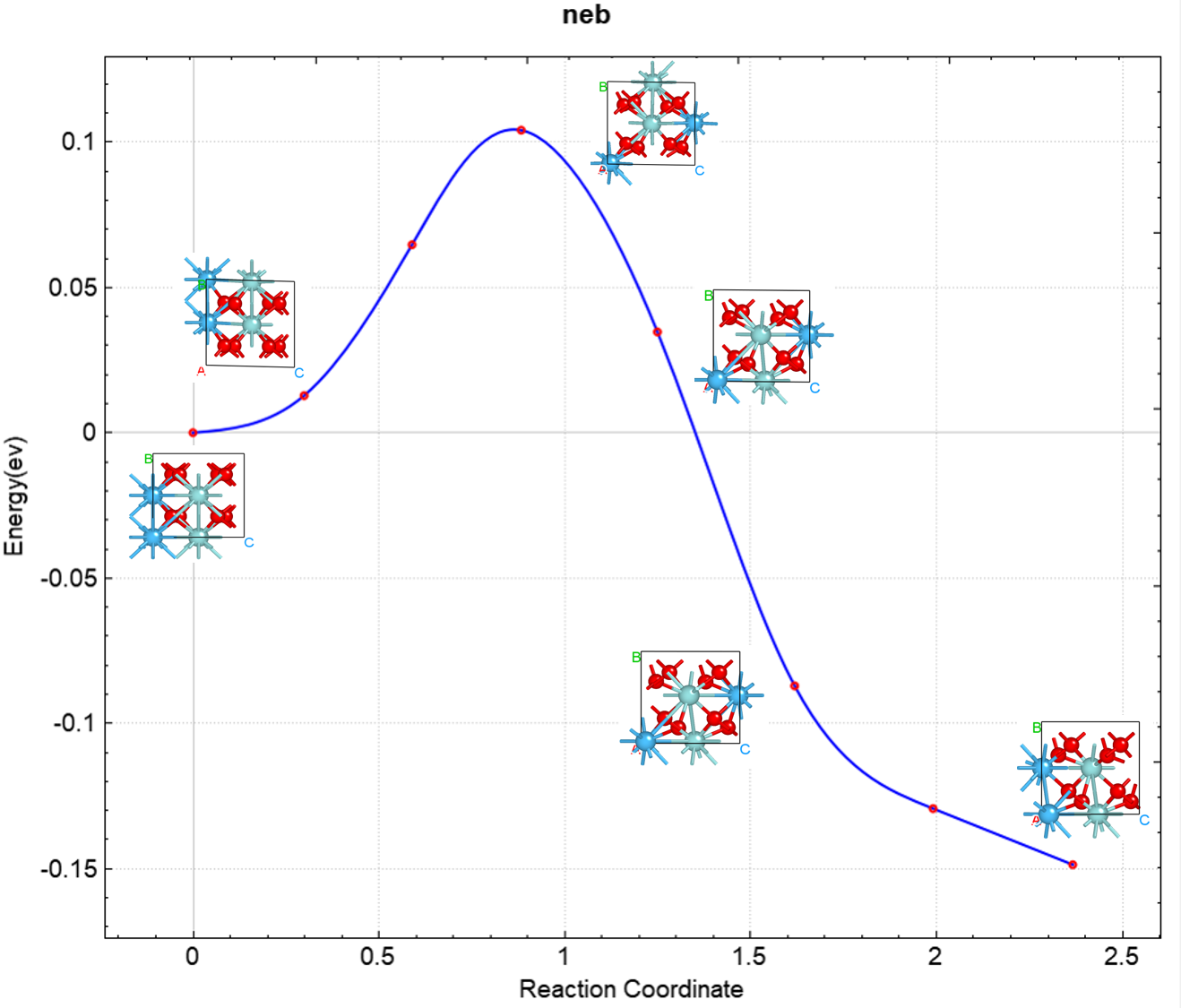
使用 Device Studio 可直接对 neb.json 文件处理出图,其操作步骤为:Simulator-->DS-PAW-->Analysis Plot,选择 neb.json 即可,可根据作图要求自定义设置面板参数。
得到的反应势垒曲线如下所示:
2.28. PBEsol泛函计算¶
本节将以 \(Ta_{2}O_{5}\) 体系为例,介绍在DS-PAW中如何使用PBEsol泛函进行弛豫计算。
2.28.1. \(Ta_{2}O_{5}\) PBEsol泛函计算输入文件¶
输入文件包含参数文件 relax.in 和结构文件 structure.as , relax.in 如下:
task = relax
sys.structure = structure.as
sys.functional = PBESOL
sys.spin = none
sys.symmetry = true
cal.ksamping = MP
cal.kpoints = [4,4,8]
cal.cutoff = 520
cal.smearing = 1
cal.sigma = 0.01
scf.convergence = 1e-5
scf.max = 60
relax.max = 60
relax.freedom = all
relax.convergence = 0.01
relax.methods = CG
relax.in 输入参数介绍:
sys.functional: 该参数控制使用泛函的类型,此例选择的泛函为PBESOL;
结构文件 structure.as 参考如下
Total number of atoms
14
Lattice
6.25000000 0.00000000 0.00000000
0.00000000 7.40000000 0.00000000
0.00000000 0.00000000 3.83000000
Cartesian
O 0.00000000 0.00000000 0.00000000
O 3.12500000 3.70000000 0.00000000
O 2.43375000 1.03970000 0.00000000
O 3.81625000 6.36030000 0.00000000
O 0.69125000 4.73970000 0.00000000
O 5.55875000 2.66030000 0.00000000
O 1.25625000 2.56114000 1.91500000
O 4.99375000 4.83886000 1.91500000
O 1.86875000 6.26114000 1.91500000
O 4.38125000 1.13886000 1.91500000
Ta 1.40250000 2.82754000 0.00000000
Ta 4.84750000 4.57246000 0.00000000
Ta 1.72250000 6.52754000 0.00000000
Ta 4.52750000 0.87246000 0.00000000
2.28.2. run程序运行¶
准备好输入文件之后,将 relax.in 和 structure.as 文件上传到服务器上运行,按照结构弛豫中介绍的方法执行 DS-PAW relax.in 。
2.28.3. analysis计算结果分析¶
根据上述的输入文件,计算完成之后将会得到 DS-PAW.log 、 relax.json 这2个文件及paw_tmp文件夹。
relax.json :弛豫计算结束得到的结构文件
将 relax.json 拖入Device Studio查看结构,或直接查看 paw_tmp 目录下的relax.tmp 文件,可得弛豫结束后晶胞常数如下表所示,与文献报道结果[6] 一致。
a (Å)
b (Å)
c (Å)
PBEsol - this work
6.190
7.315
3.786
PBEsol - ref
6.197
7.333
3.793