4. 支持的文件类型¶
Device Studio支持的文件类型有 .hzw、.xyz、.cif、.dsxml、.pdb、.mol、.xsd、scf.input、.py、POSCAR、CONTCAR 等,其中 .hzw 为Device Studio特有文件格式。Device Studio可根据用户需要导出 .hzw、.xyz 格式的结构文件,并可将结构文件的3D可视化结果以 .png 图片格式导出;对于晶体结构,可识别其空间群结构信息,并导出 .cif 格式的结构文件。
5. 结构建模¶
Device Studio支持从本地或在线数据库导入结构,或在导入结构的基础上搭建新的结构,支持创建各种分子、晶体和LCR/LR/FCB/FB/BCT/BT等典型的器件结构。Device Studio可根据用户提出的匹配精度要求,自动劈裂晶面,并进行匹配搭建器件结构,可以自动匹配搭建多层膜器件或晶体结构。
5.1. 导入结构¶
导入结构前需要创建新项目或打开已建项目,在存在项目的基础上导入结构。创建项目的操作为:双击Device Studio图标快捷方式登录并启动软件,其图形界面如图5.1-1所示,根据界面提示选择创建一个新的项目( Create a new Project )或打开一个已经存在的项目( Open an existing Project )的按钮,选中之后点击界面中的 OK 按钮即可。若选择创建一个新的项目,用户可根据需要给该项目命名,如本项目命名为 DeviceStudio。
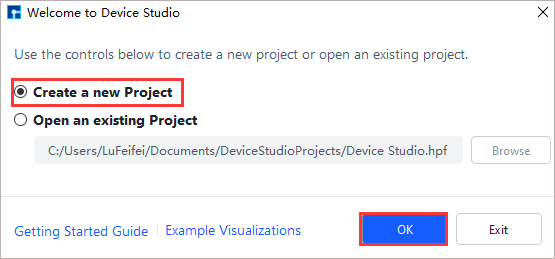
图5.1-1: 启动软件后选择创建或打开项目的图形界面¶
5.1.1. 本地数据库导入结构¶
Device Studio自带本地数据库,该数据库包含500多种常用或热门材料,后期将会不断更新壮大数据库,若用户需大量级数据库,可关注鸿之微软件 FIRST ,该软件内置了三大数据库QuickMol、QuickCrystal和ACED,材料条目超过 1000万。用户可通过本地数据库导入结构,如导入ZnO-MgO-Si器件结构,其导入结构的操作如图5.1-2红色部分所示:File → Import → Import Local,之后弹出图形界面如图5.1-3所示,根据界面提示在本地数据库中找到ZnO-MgO-Si所在的文件夹,选中ZnO-MgO-Si结构文件,点击图5.1-3中的打开按钮即可导入ZnO-MgO-Si结构,导入ZnO-MgO-Si结构后的图形界面如图5.1-4所示,其中 ZnO-MgO-Si.hzw 结构文件在 Project Explorer(项目管理)区域,结构的3D视图显示在3D Viewer(3D显示)区域。
若不想通过数据库导入结构,知道结构文件的位置,如ZnO-MgO-Si结构,直接鼠标左击选中该结构文件,拖动到Device Studio的Project Explorer(项目管理)区域即可导入该结构,同时其结构的3D视图显示在3D Viewer(3D显示)区域。

图5.1-2: 弹出本地数据库导入结构操作界面¶
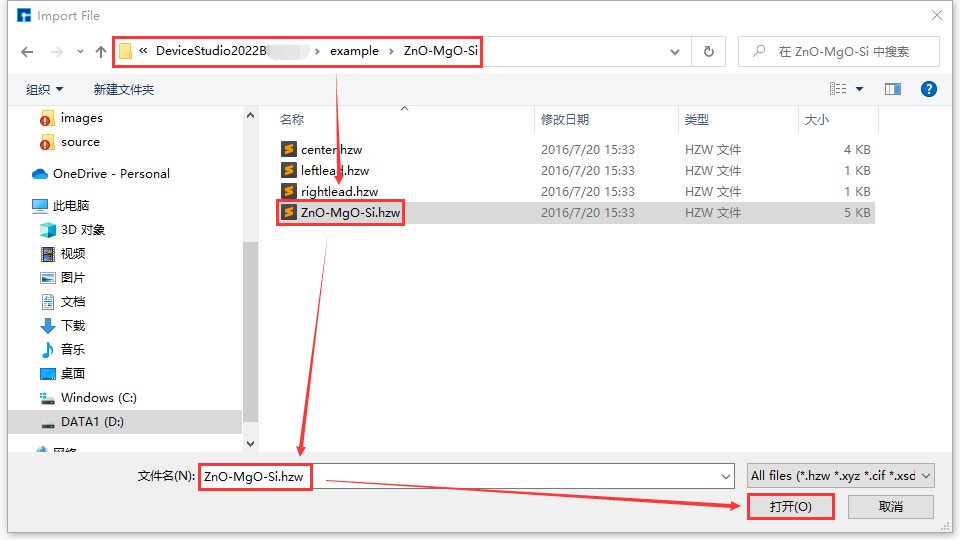
图5.1-3: 选中ZnO-MgO-Si结构文件界面¶
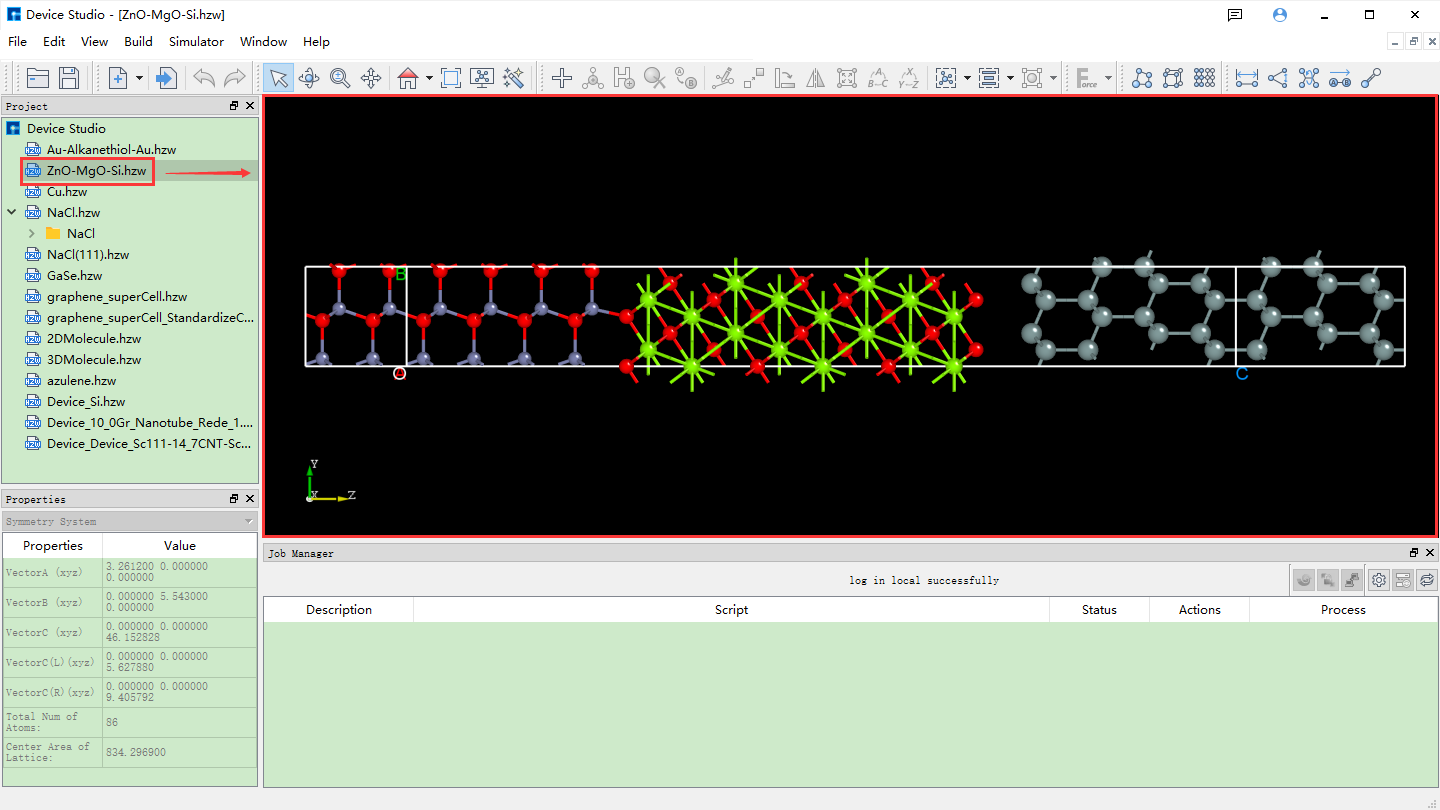
图5.1-4: 导入ZnO-MgO-Si结构后的Device Studio界面¶
5.1.2. 在线数据库导入结构¶
Device Studio支持连接在线数据库Materials Project,如图5.1-5红色部分所示:点击 File → Import → Import Online 则弹出从Materials Project数据库导入结构的界面,在该界面中用户可通过输入元素、化学式或mp序号的方式来搜索原子结构。
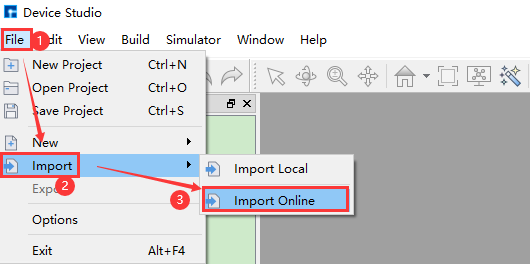
图5.1-5: 弹出在线数据库导入结构操作界面¶
如图5.1-6所示,通过输入元素来搜索结构,输入Si元素,点击 Search 按钮或点击键盘 Enter 键,则出现很多只含有Si元素的原子结构,如Si4结构,左边为其详细的化学式,右边为其对应的空间群结构,选中之后可在界面左下角红色框区域查看该结构的化学式、空间群对称信息以及原子坐标等详细信息;可在界面右下角红色框区域查看到该结构的3D显示。对于结构的3D显示,可通过滚动鼠标中键将3D显示结果放大或缩小;可鼠标右键选中该结构,通过拖动鼠标来将3D视图显示结果进行旋转,确定好搜索的原子结构后点击界面中的 Add 按钮则可将该原子结构导入,其结构文件保存在软件的项目管理区域(Project Explorer),同时可在3D显示区域查看到该原子结构的3D视图显示,通过在线数据库Materials Project导入Si4结构后的Device Studio界面如图5.1-7所示。
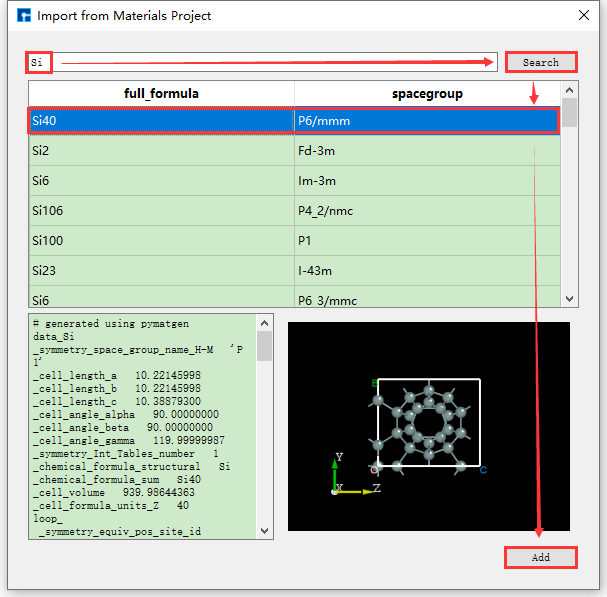
图5.1-6: Materials Project元素搜索并导入结构的操作界面¶
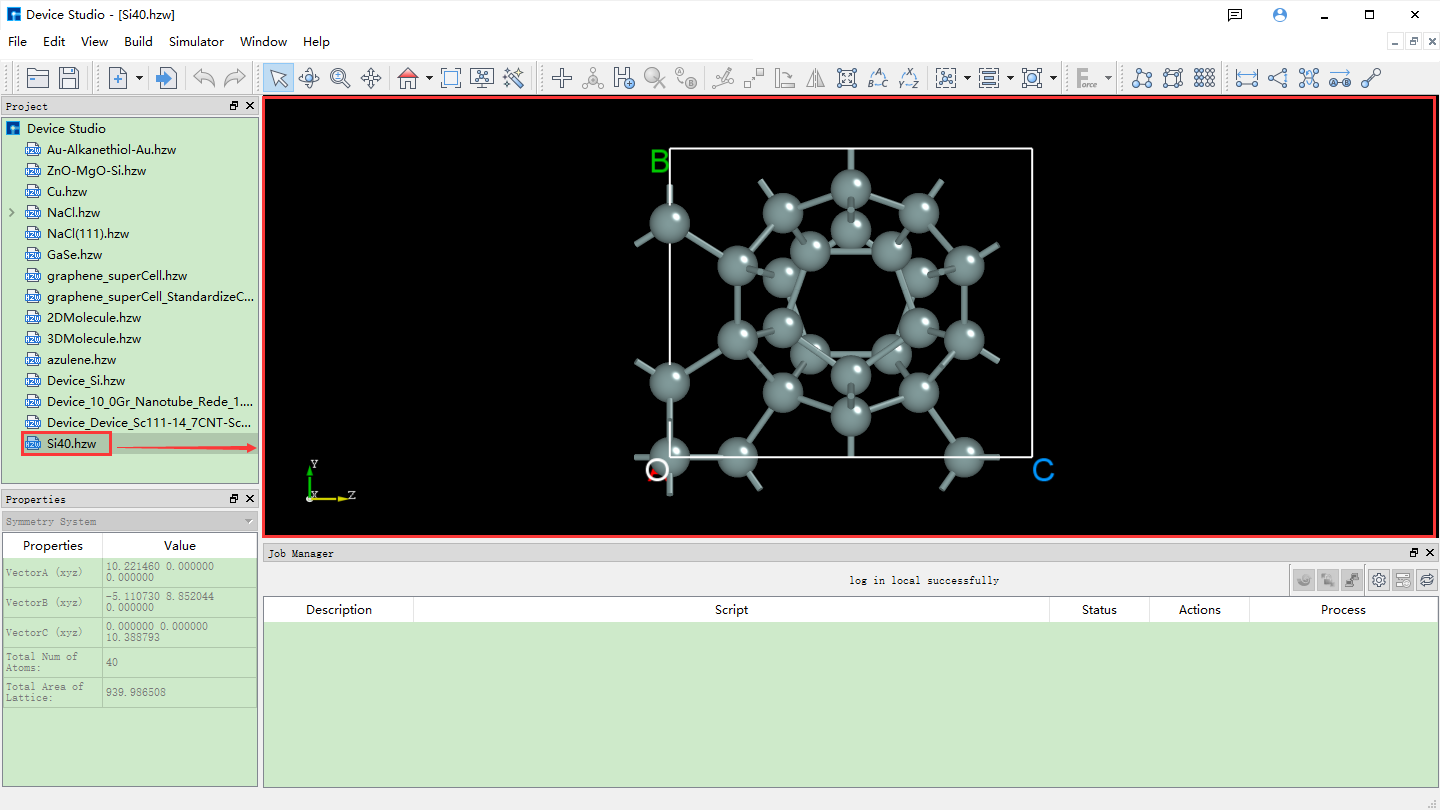
图5.1-7: 导入Si4结构后的Device Studio界面¶
通过化学式、mp序号来搜索并导入结构的操作与通过元素搜索并导入结构的操作一致,其操作界面分别如图5.1-8、5.1-9所示。
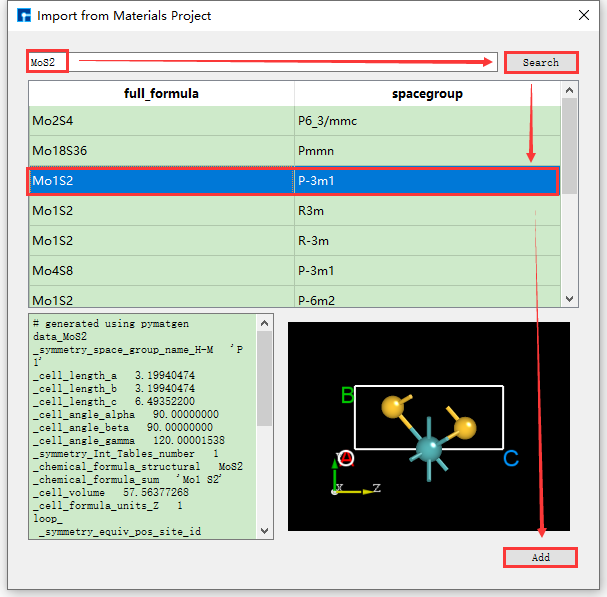
图5.1-8: Materials Project化学式搜索并导入结构的操作界面¶
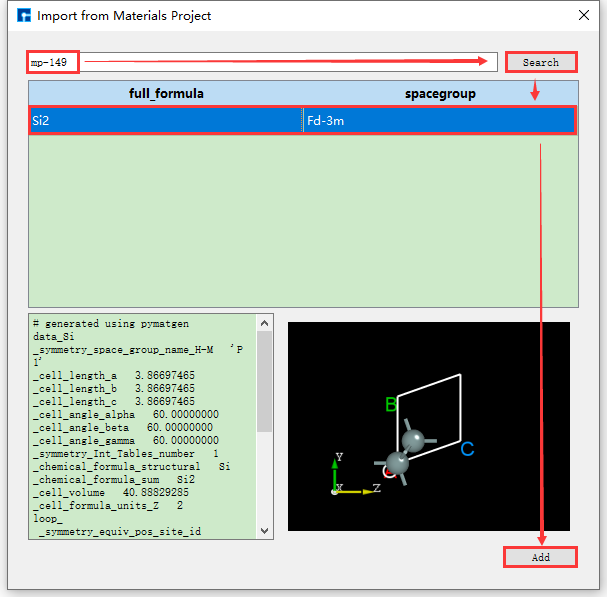
图5.1-9: Materials Project mp序号搜索并导入结构的操作界面¶
5.2. 2D分子建模¶
Device Studio 2022A 版新增 2D分子建模 功能,在2D分子建模的界面中,其工具栏包含绘制分子结构式的多种键和工具,包括化学键、环形模板、折叠链、船式结构、椅式结构、箭头、边框以及文本编辑工具等,用户可通过该功能快捷方便的绘制各种2D平面分子结构式和编写方程式,并支持将2D分子结构转化为3D分子结构。
Device Studio具有对2D分子结构进行编辑的功能,用户可对分子结构进行旋转、修改键宽、键长、键位颜色、键位类型、键位属性转换、加氢、减氢、加电荷、减电荷等操作。
以搭建3个相连苯环的2D分子结构并将其转换为对应的3D分子结构为例,其搭建步骤如图5.2-1所示。
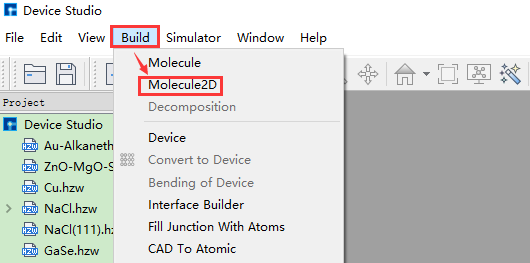
(a): 弹出2D分子建模界面操作¶
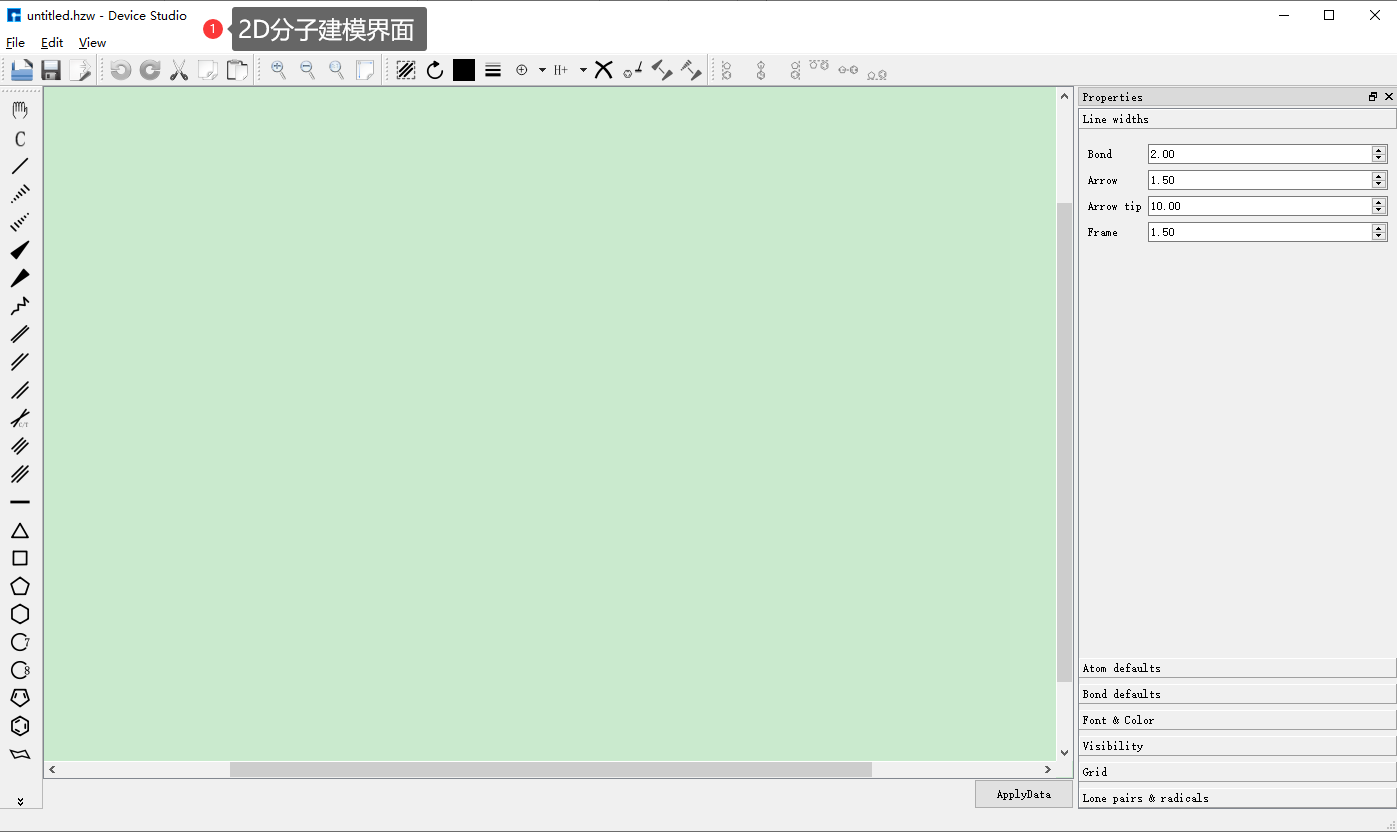
(b): 2D分子建模界面¶
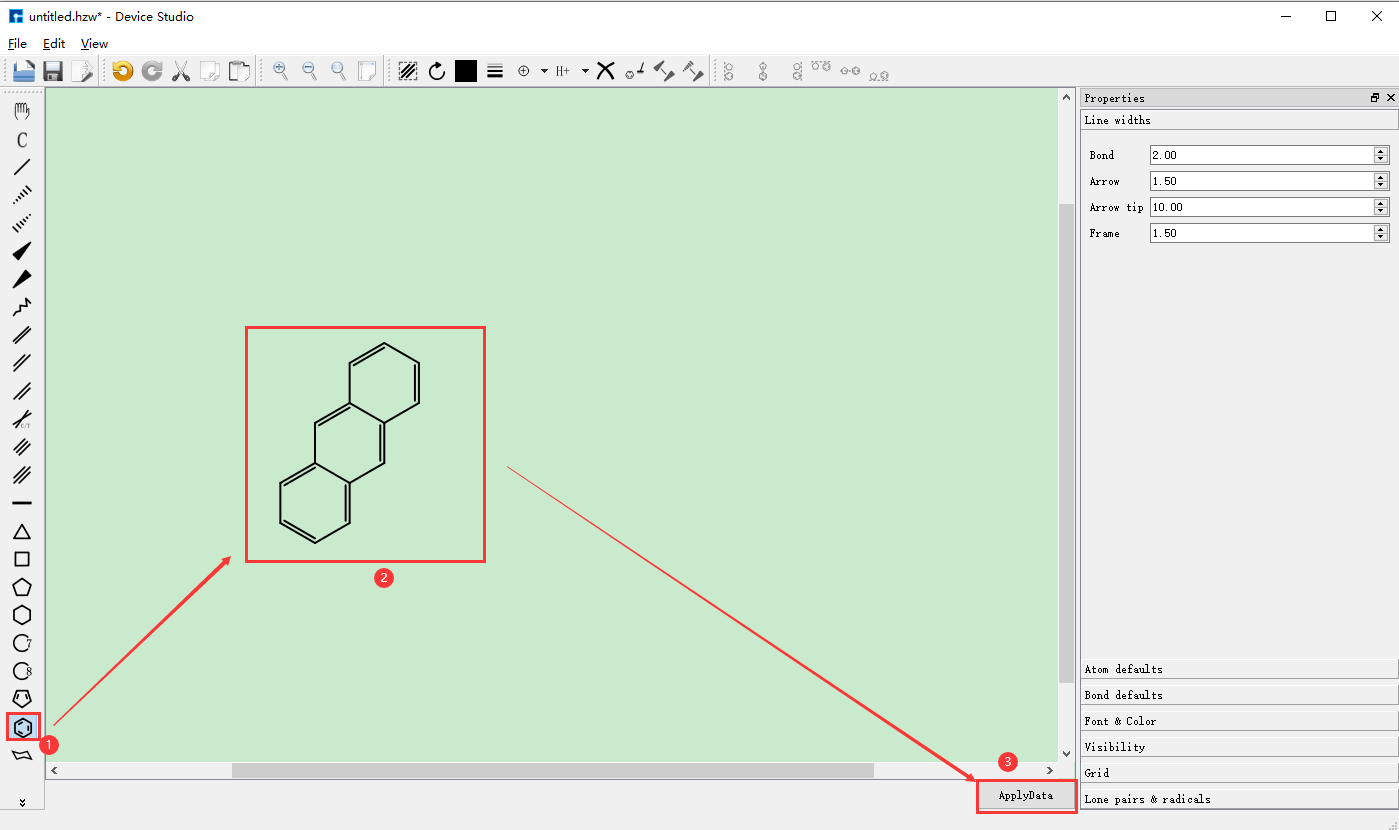
(c): 搭建2D分子结构并转换为3D分子结构操作¶
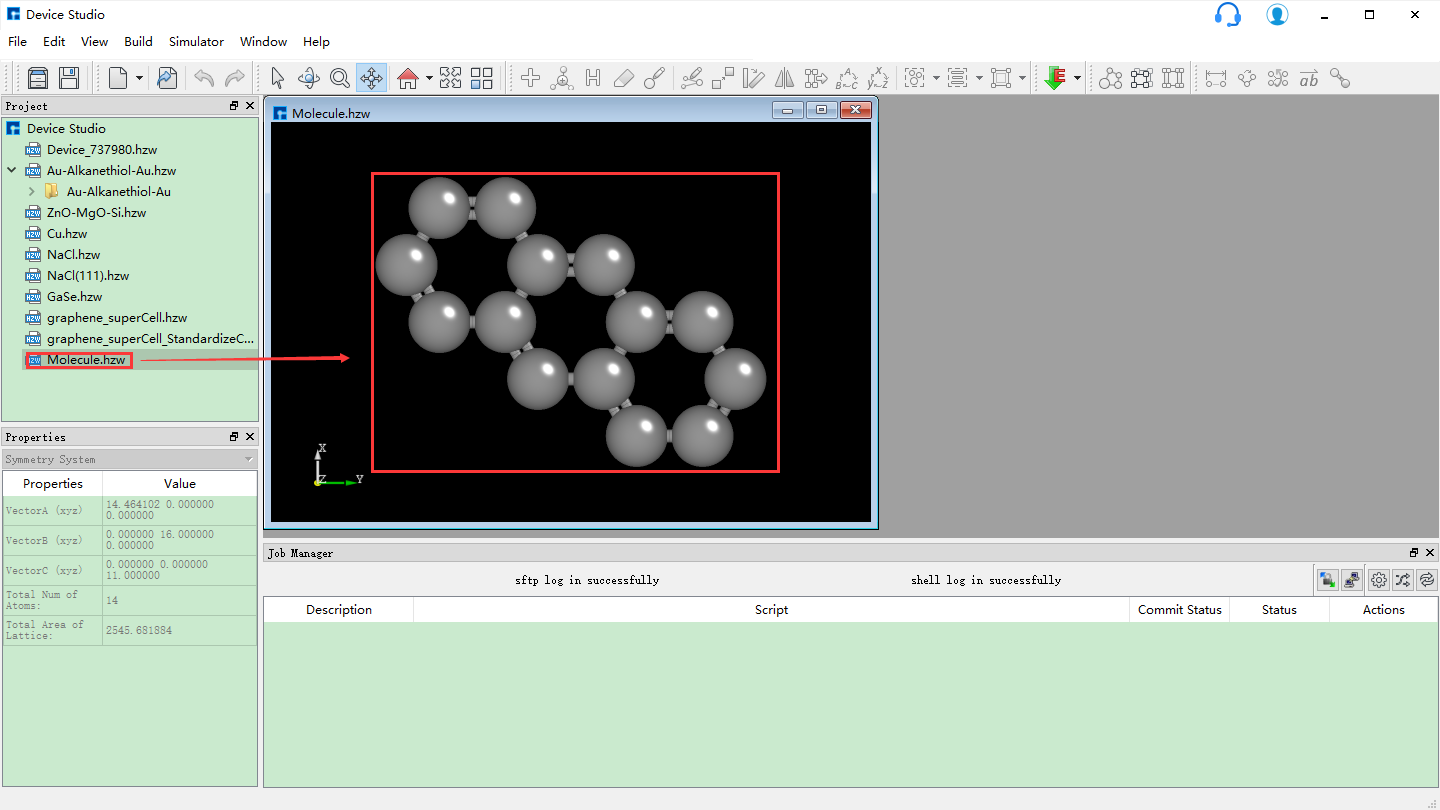
(d): 2D分子结构转换为3D分子结构后的Device Studio界面¶
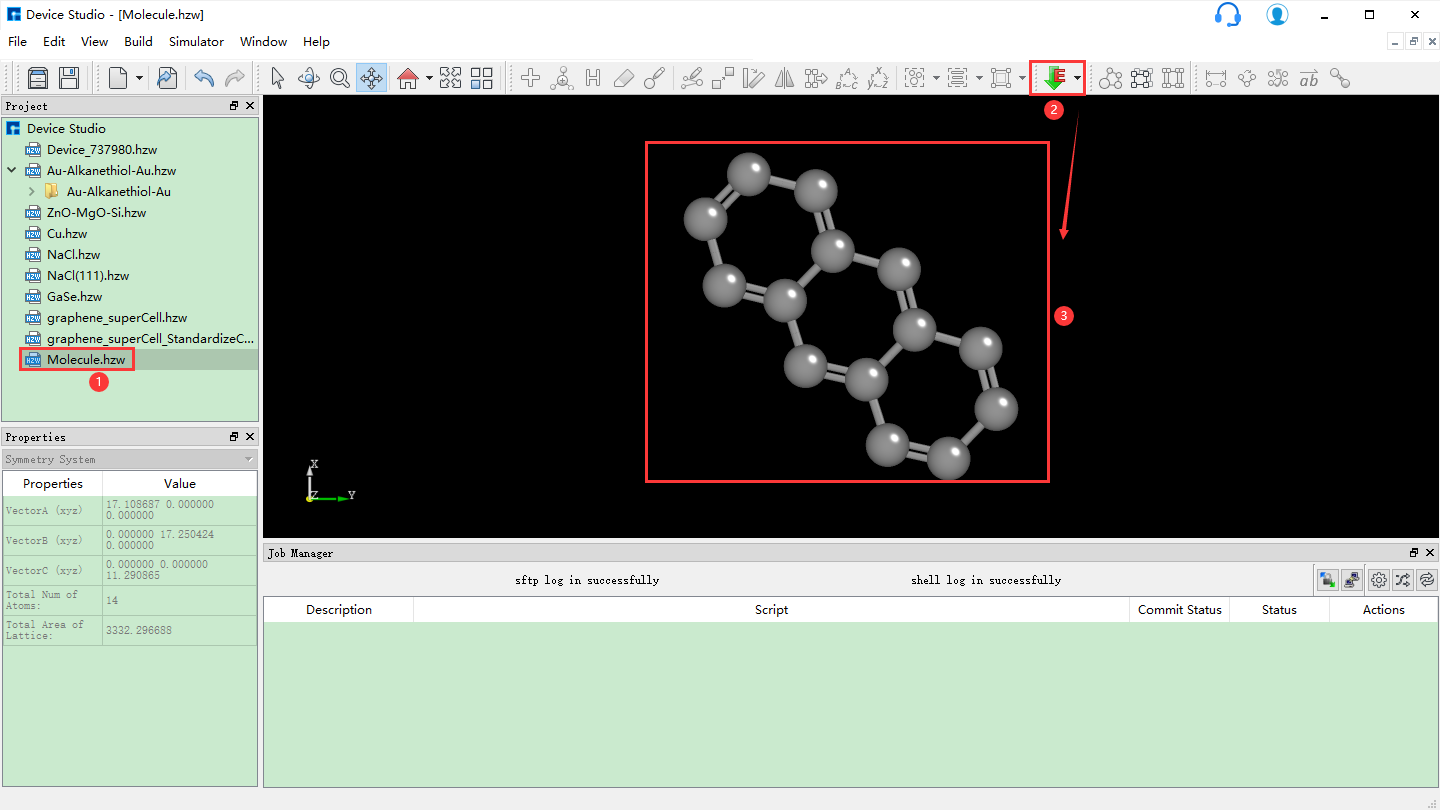
5.3. 3D分子建模¶
Device Studio 2022A 版新增 3D分子建模 功能,用户可通过该功能方便快捷的搭建3D分子结构,Device Studio支持对3D分子结构进行编辑,并可查看分子结构的原子坐标和原子之间成键的详细信息,可对分子结构进行共键调整、原子夹角调整、二面角调整、加氢、平移、旋转、重命名、复制片段、增、删、改等一系列操作。Device Studio搭建3D分子结构的操作界面如图5.3-1所示。
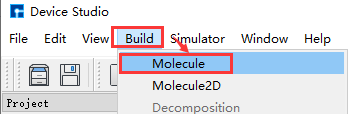
(a): 弹出3D分子建模界面操作¶
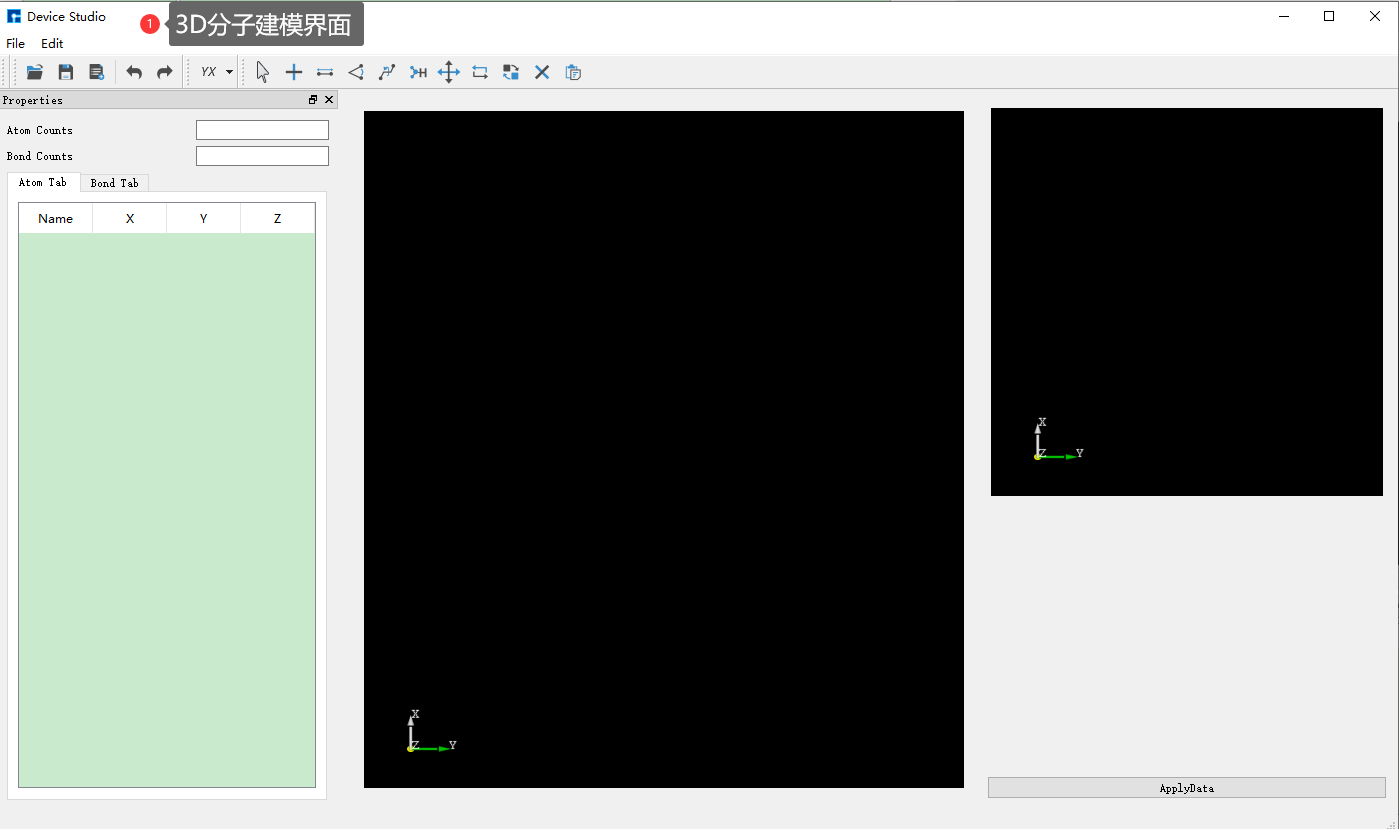
(b): 3D分子建模界面¶
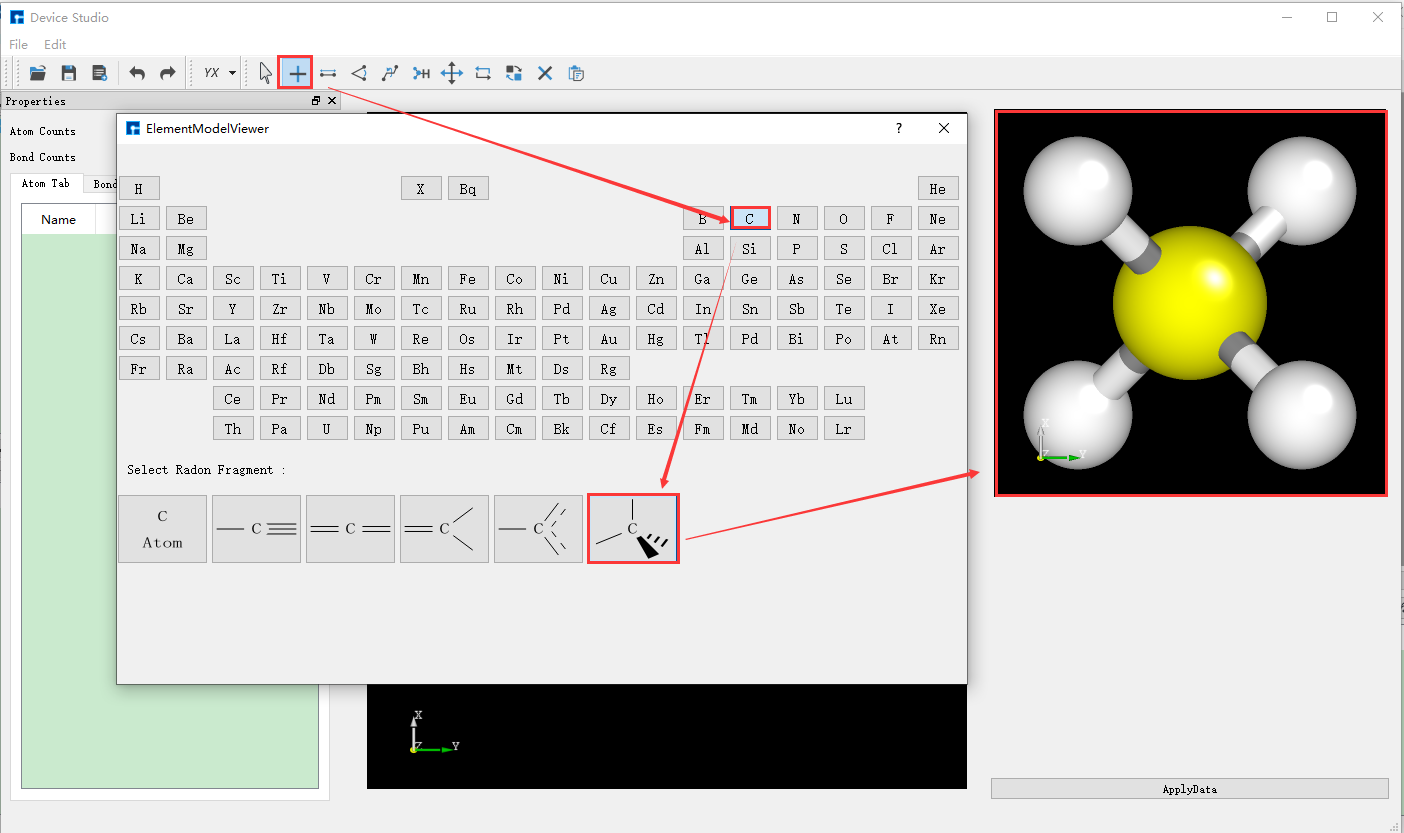
(c): 搭建3D分子结构操作一¶
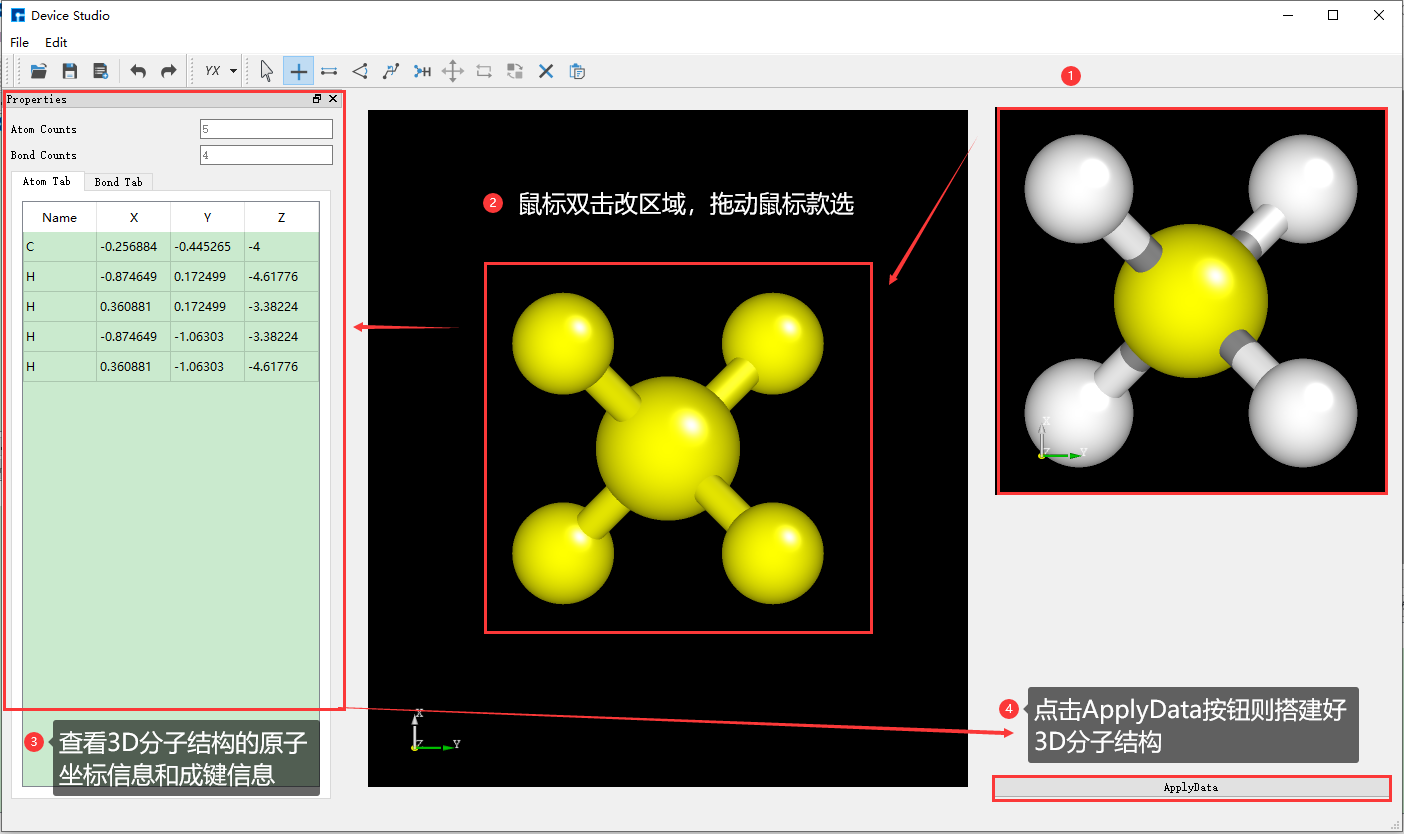
(d): 搭建3D分子结构操作二¶
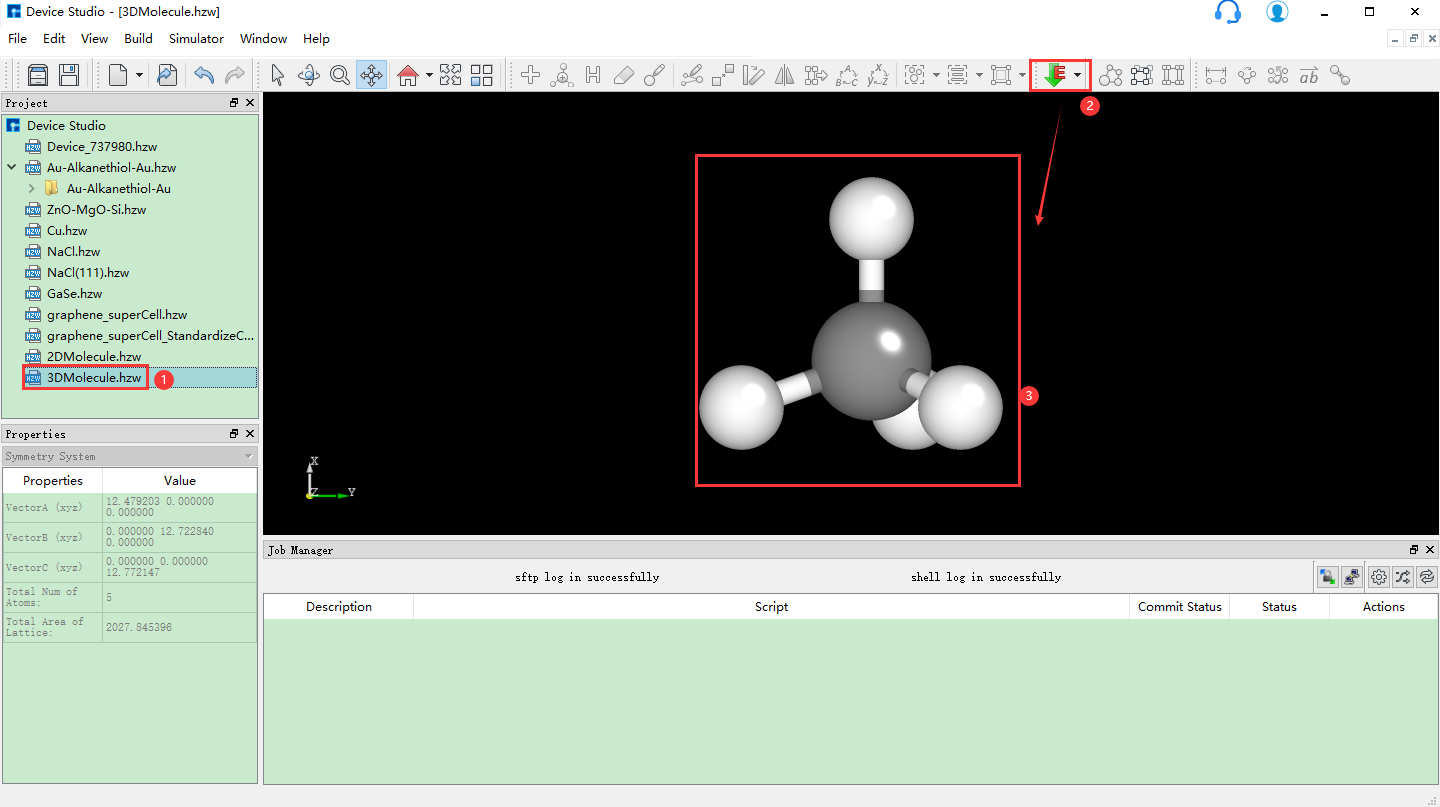
5.4. 晶体建模¶
对于晶体建模,用户可先在本地数据库或在线数据库搜索该结构是否存在,若存在,直接导入;若不存在,可自行构建,或导入一个结构,在其基础上进行构建,具体如何构建晶体结构,用户可根据需要自行选择。
以构建NaCl晶体结构为例,在如图5.1-7所示的界面中点击 Build → Crystal ,弹出构建晶体结构的界面如图5.4-1所示。
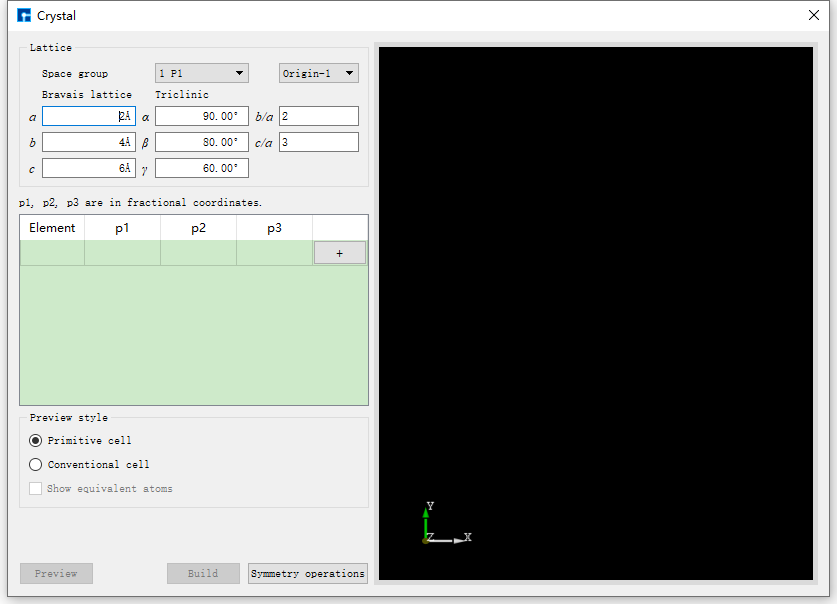
图5.4-1: 构建晶体结构界面¶
若不知道NaCl晶体的空间群信息,在如图5.4-1所示的界面中构建NaCl晶体结构,则可按照图5.4-2红色框选部分及箭头指向所示,一步步填写或选中对应信息,点击 Preview 按钮在界面右侧区域预览所搭建的结构,该步骤主要用于检查搭建的结构是否正确,是否是所需结构,之后点击 Build 则搭建好NaCl晶体结构,其结构文件保存在软件的项目管理区域(Project Explorer),同时可在3D显示区域查看到该结构的3D视图;反之,若在预览过程中发现所搭建的结构不正确,则可在图5.4-1所示界面中重新搭建。搭建结构过程中,用户可根据需要选择界面中的 Display Style,即选中 Primitive 或 Conventional ;选择是否显示等价位置原子,即是否勾选 Show equivalent atoms。
- NaCl晶体结构的p1、p2、p3值如下表所示:
Element
p1
p2
p3
Na
0
0
0
Na
0.5
0.5
0
Na
0.5
0
0.5
Na
0
0.5
0.5
Cl
0.5
0
0
Cl
0
0.5
0
Cl
0
0
0.5
Cl
0.5
0.5
0.5
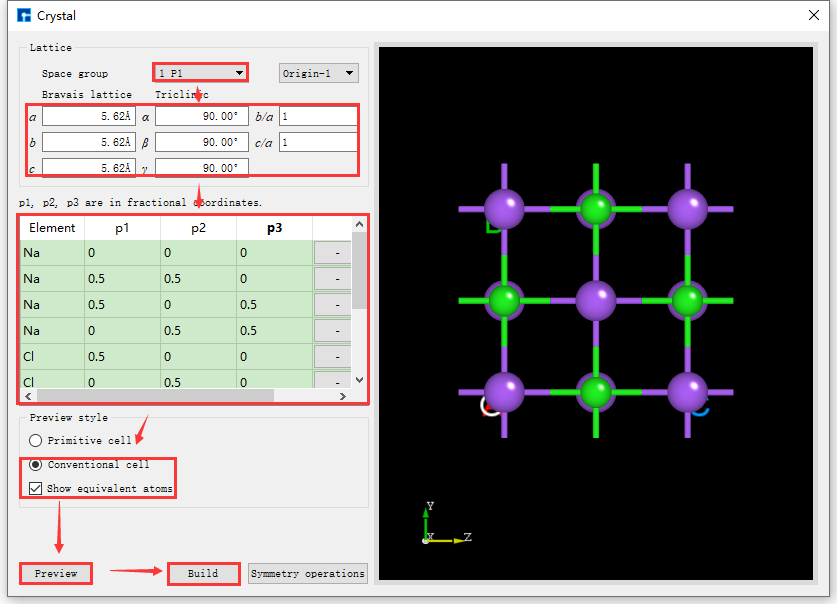
图5.4-2: 无空间群信息直接构建NaCl晶体操作界面¶
若知道所搭结构的空间群信息,可更方便快捷的搭建结构,Device Studio支持261种空间群(包含230种空间群和31种扩展子群)的选择。如NaCl晶体的空间群为 225 FM-3M,知道该信息,在如图5.4-1所示的界面中构建NaCl晶体,则可按照图5.4-3红色框选部分及箭头指向所示填写对应信息进行搭建,其余操作与上述不知空间群信息一致,这里不做详细说明。对比图5.4-2与图5.4-3可知,知道空间群信息可以更简便的搭建晶体结构。
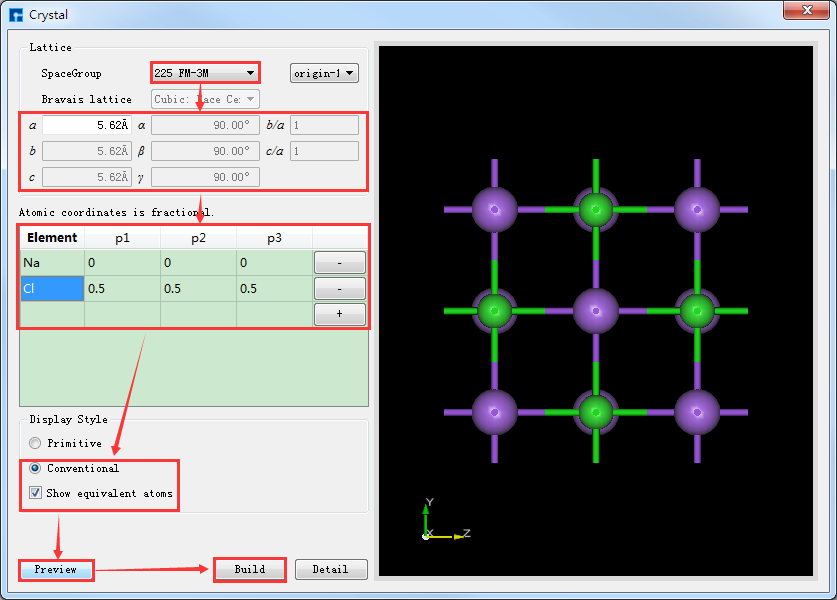
图5.4-3: 根据空间群信息构建NaCl晶体操作界面¶
图5.4-2与图5.4-3均为搭建NaCl晶体结构时,选择显示等价位置原子的结果,即勾选 Show equivalent atoms,若不勾选,其操作界面如图5.4-4所示,点击界面中的 Build 按钮则搭建好NaCl晶体结构,其结构文件保存在软件的项目管理区域,同时其结构的3D视图显示在3D显示区域,搭建好NaCl晶体结构的界面如图5.4-5所示。
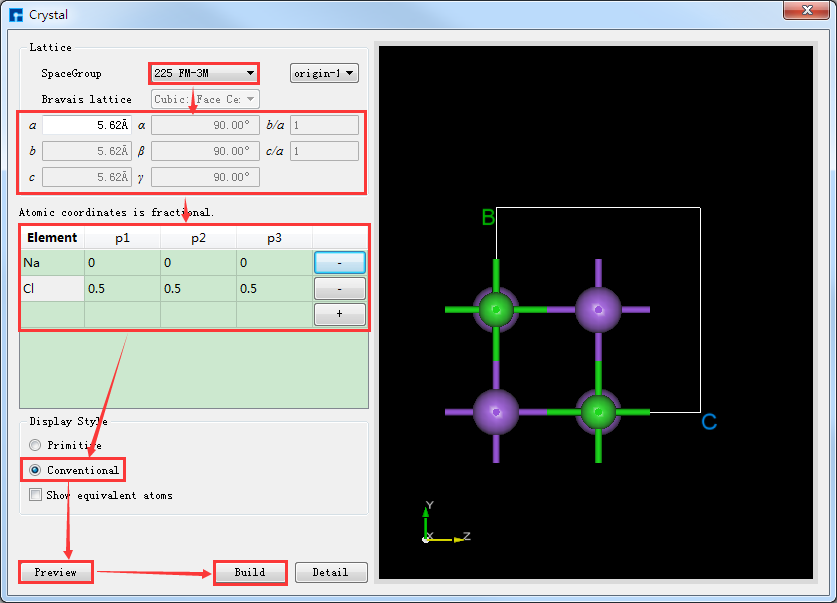
图5.4-4: 不显示等价位置原子构建NaCl晶体操作界面¶
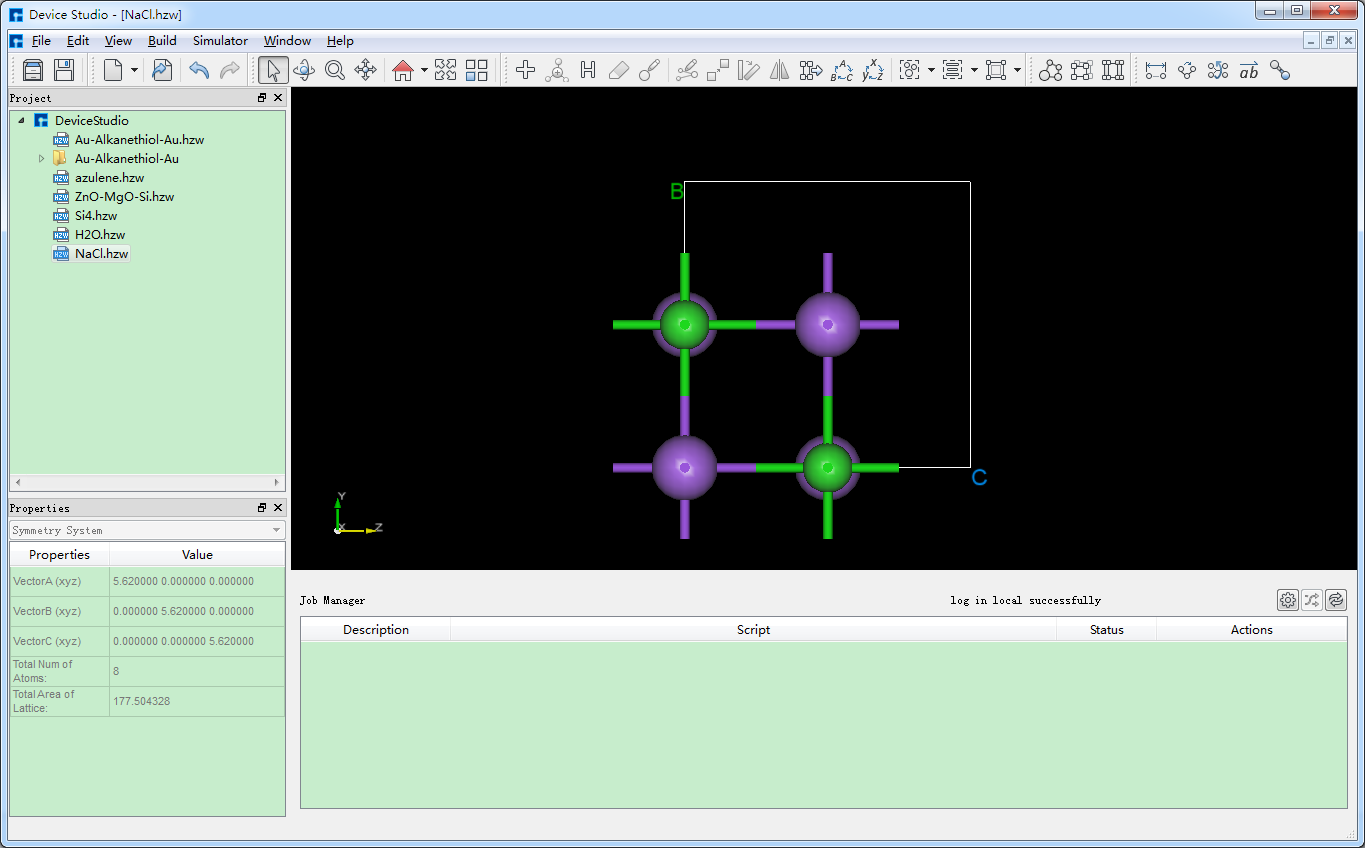
图5.4-5: 搭建好NaCl晶体结构的Device Studio界面¶
5.5. 晶体结构的切面/切片¶
晶体结构的切面/切片,该功能仅在存在晶体结构的基础上进行。以在5.4节中搭建的NaCl晶体结构的基础上进行切面/切片搭建Thickness为9.73埃的NaCl(1 1 1)晶体结构为例,在如图5.4-5所示界面中点击 Build → Surface/Slab ,弹出将NaCl晶体结构进行切面/切片的界面如图5.5-1所示。
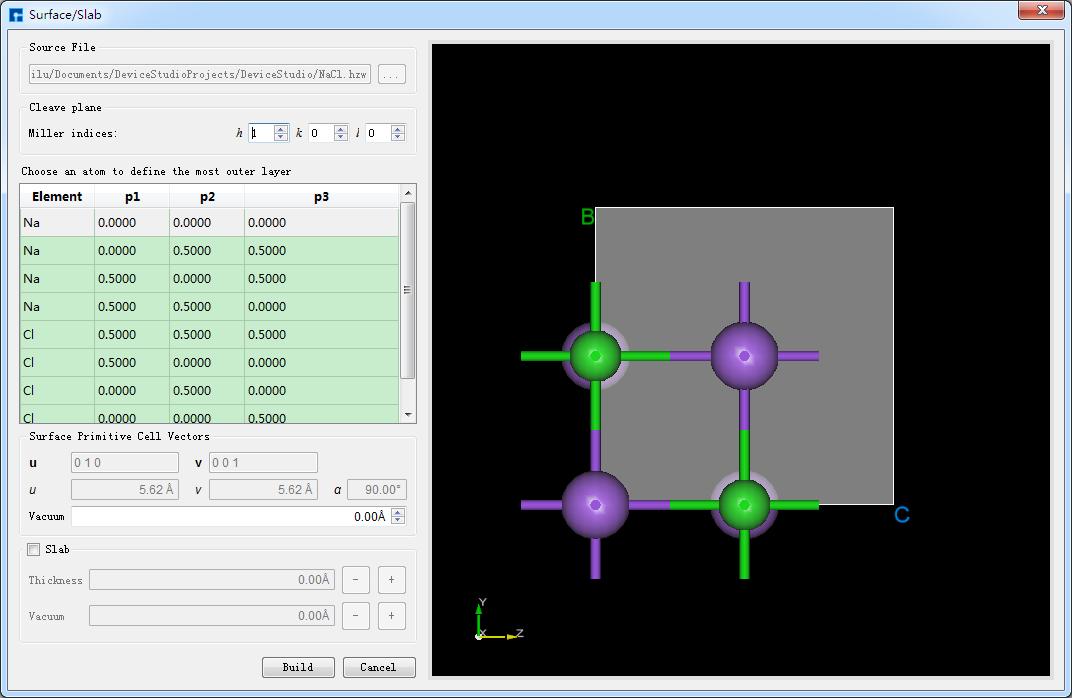
图5.5-1: 将NaCl晶体结构进行切面/切片的界面¶
根据需要按照图5.5-2中红色框选部分及箭头指向所示,在图5.5-1所示界面中设置Miller指数(1 1 1),并在下拉表格中选定Na(0.0000 0.0000 0.0000)原子为切面的起始原子,勾选 Slab ,点击Thickness后的“+”号将Thickness设置为9.73埃,同时可在界面右侧区域预览进行切面/切片的结构,之后点击 Build 则搭建好Thickness为9.73埃的NaCl(1 1 1)晶体结构,同时其结构文件挂载在Device Studio的项目管理区域,3D视图显示在Device Studio的3D显示区域,搭建好Thickness为9.73埃的NaCl(1 1 1)晶体结构的Device Studio界面如图5.5-3所示。
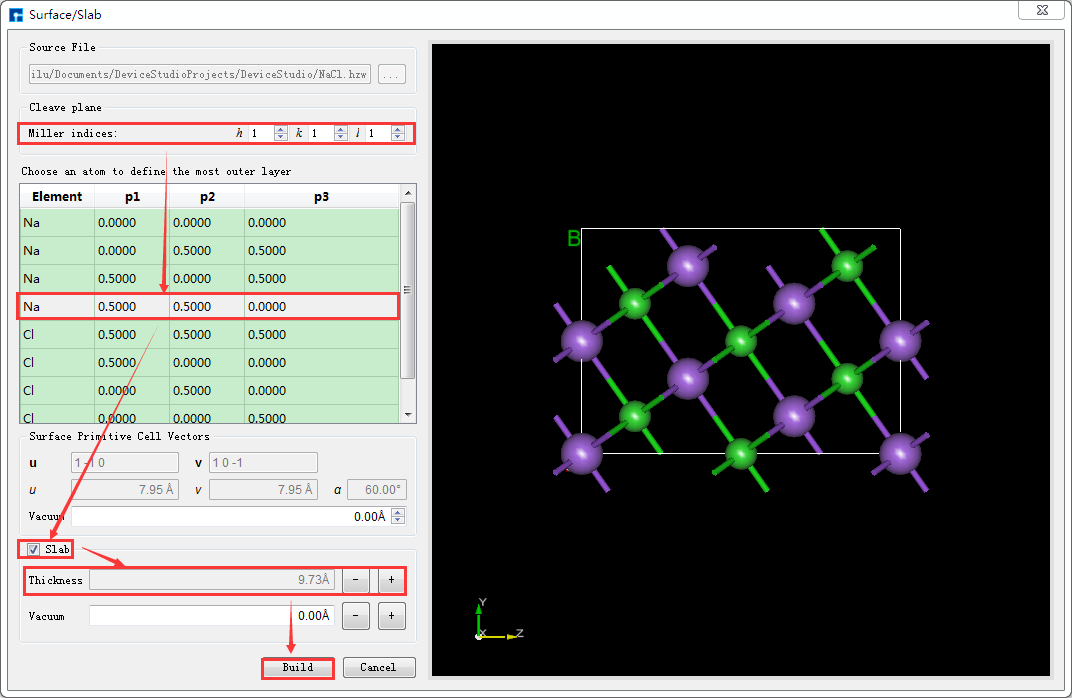
图5.5-2: 搭建Thickness为9.73埃的NaCl(1 1 1)晶体结构的操作界面¶
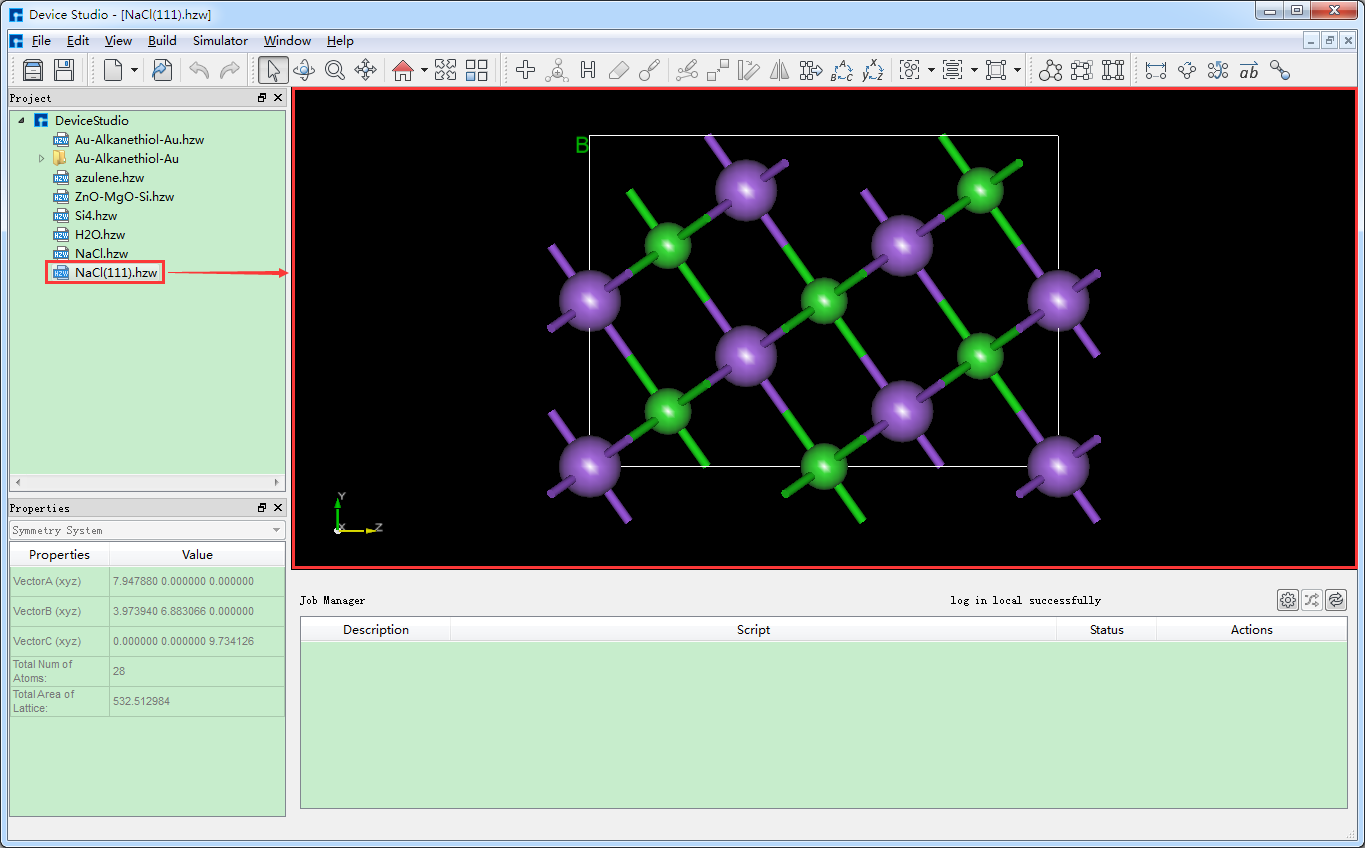
图5.5-3: 搭建好Thickness为9.73埃的NaCl(1 1 1)晶体结构的Device Studio界面¶
5.6. 晶体结构的晶胞重定义¶
在Device Studio中导入Cu原胞结构后的界面如图5.6-1所示,点击界面中的 Build → Redefine Crystal,弹出将Cu原胞结构晶体重定义界面如图5.6-2所示,按照图5.6-3中的红色框选部分填写参数,填写好参数后,点击 Preview 按钮则可在界面右侧预览扩胞后的结构,之后点击 Build 按钮则将Cu原胞扩为单胞,同时其结构文件 Cu_Rede.hzw 挂载在Device Studio的项目管理区域,结构的3D视图在Device Studio的3D显示区域如图5.6-4所示。
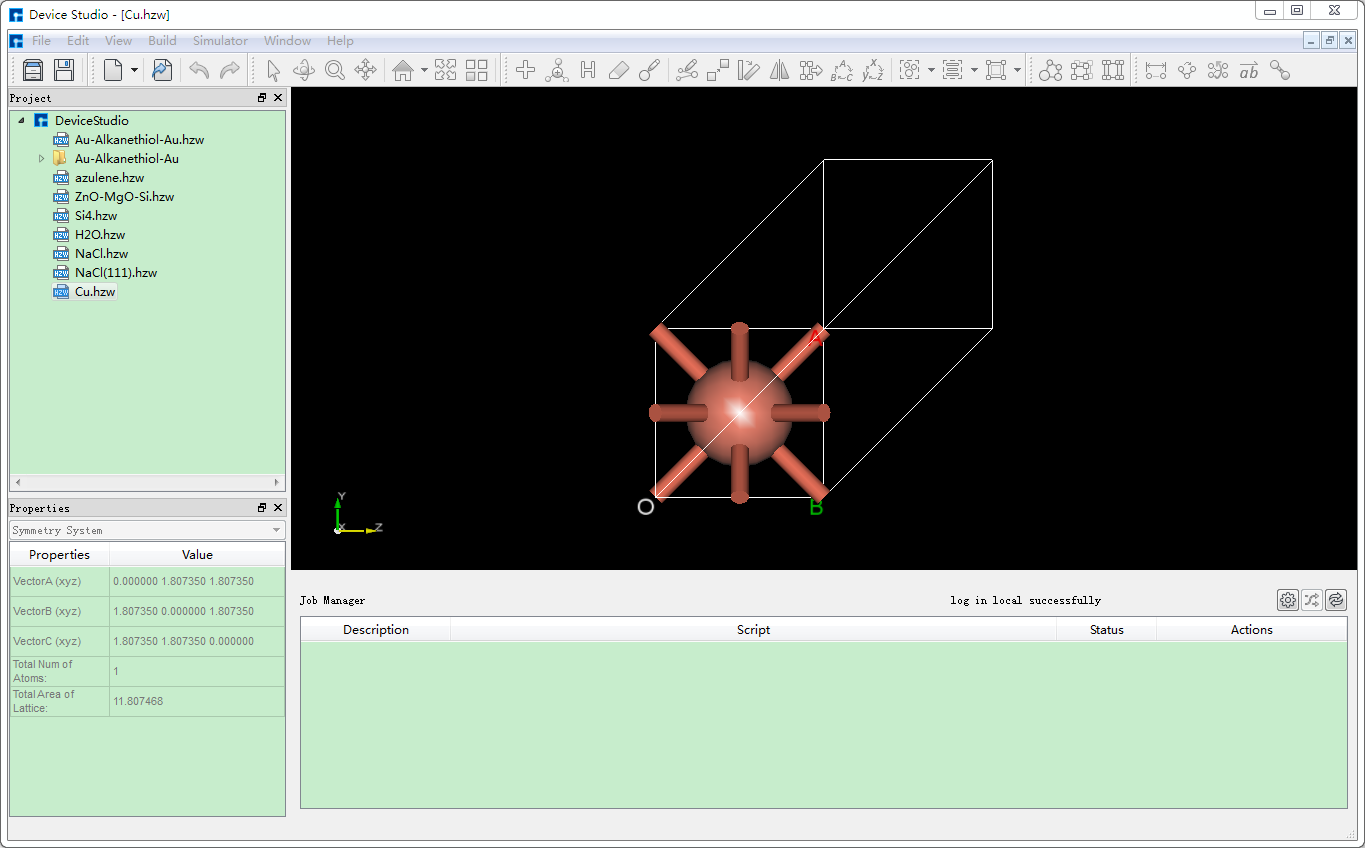
图5.6-1: 导入Cu原胞结构( Cu.hzw )后的Device Studio界面¶
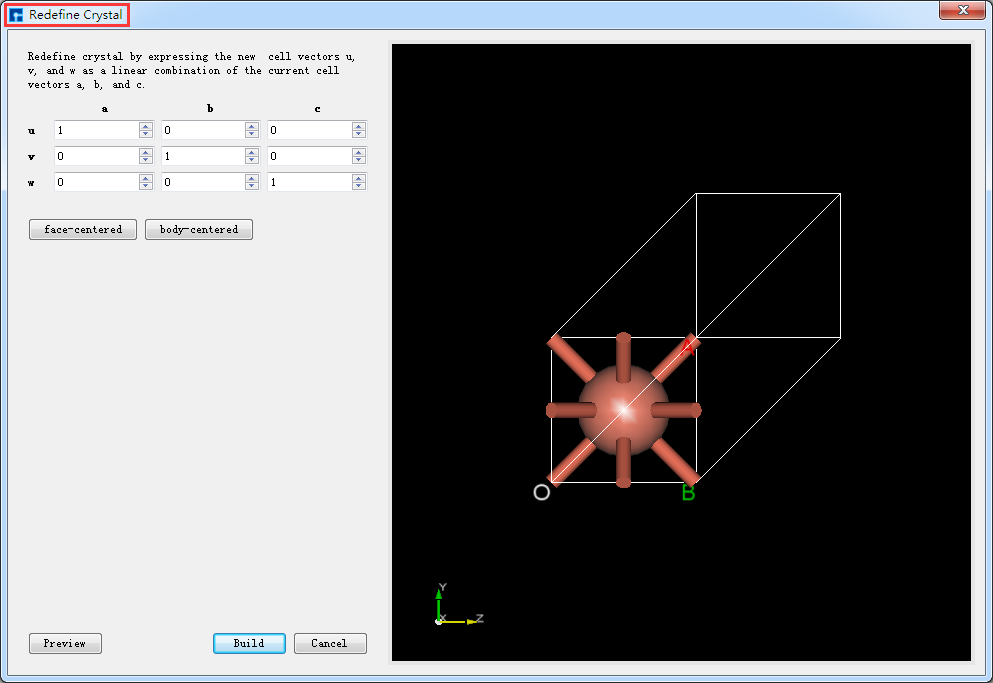
图5.6-2: 将Cu原胞结构晶胞重定义界面¶
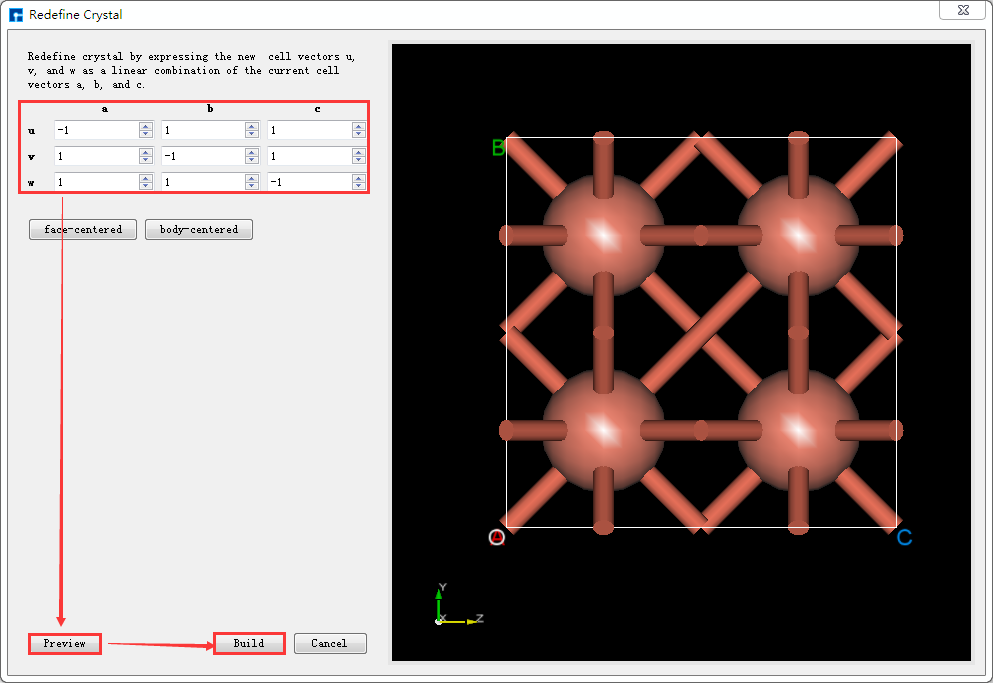
图5.6-3: 将Cu原胞结构扩为单胞的操作界面¶
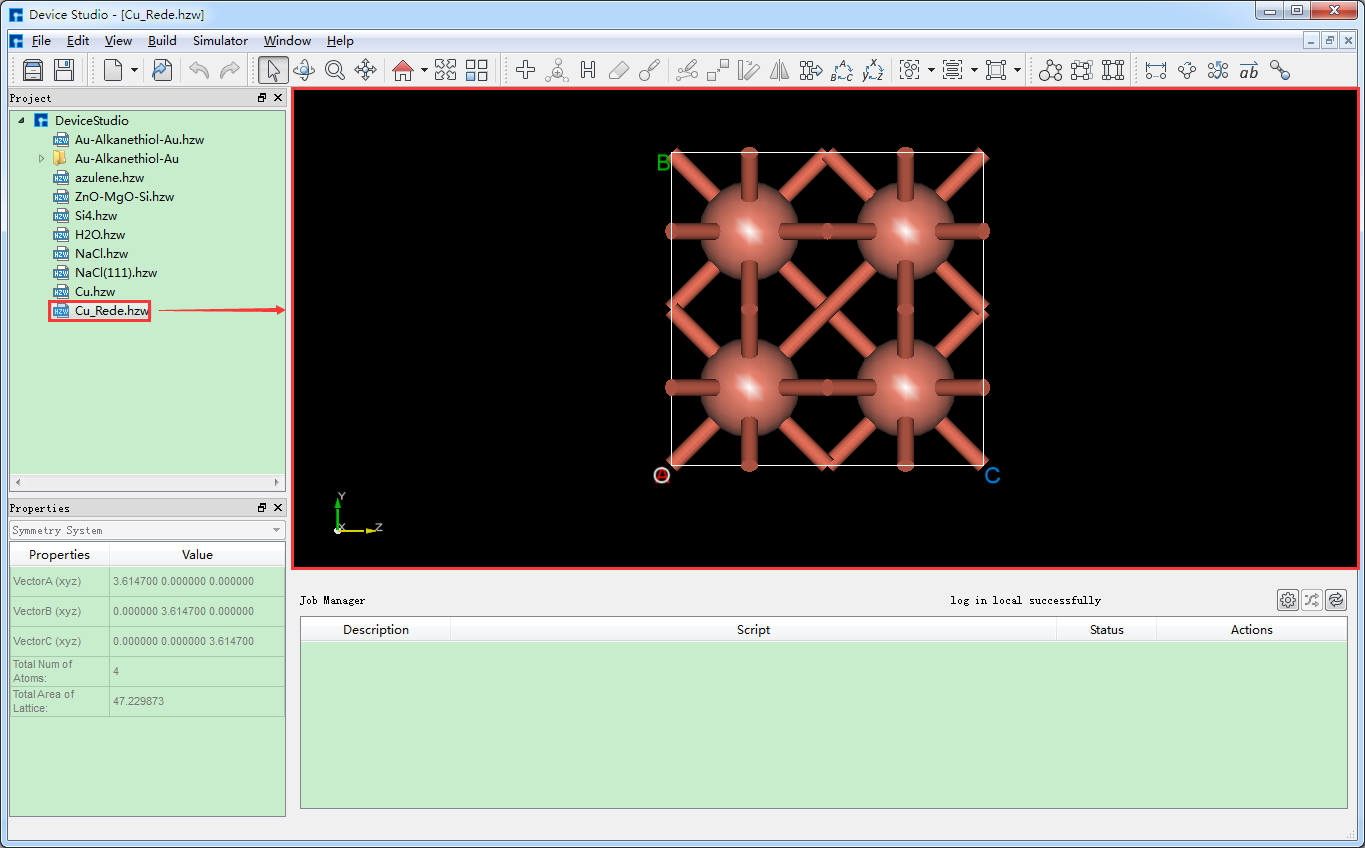
图5.6-4: Cu的单胞( Cu_Rede.hzw )结构在Device Studio中的3D显示¶
运用Device Studio的晶体结构的晶胞重定义功能可以将单胞扩为超胞,其操作与将Cu原胞扩为单胞的操作一致,以将Cu单胞扩为4*4*4的超胞为例,点击图5.6-4中的 Build → Redefine Crystal,弹出将Cu单胞结构晶胞重定义界面,其操作界面如图5.6-5所示,按照图5.6-5中的红色框选部分及箭头指向所示一步步操作,之后点击 Build 按钮则将Cu单胞扩为4*4*4的超胞,同时其结构文件 Cu_Rede_Rede.hzw 挂载在Device Studio的项目管理区域,结构的3D视图在Device Studio的3D显示区域如图5.6-6所示。
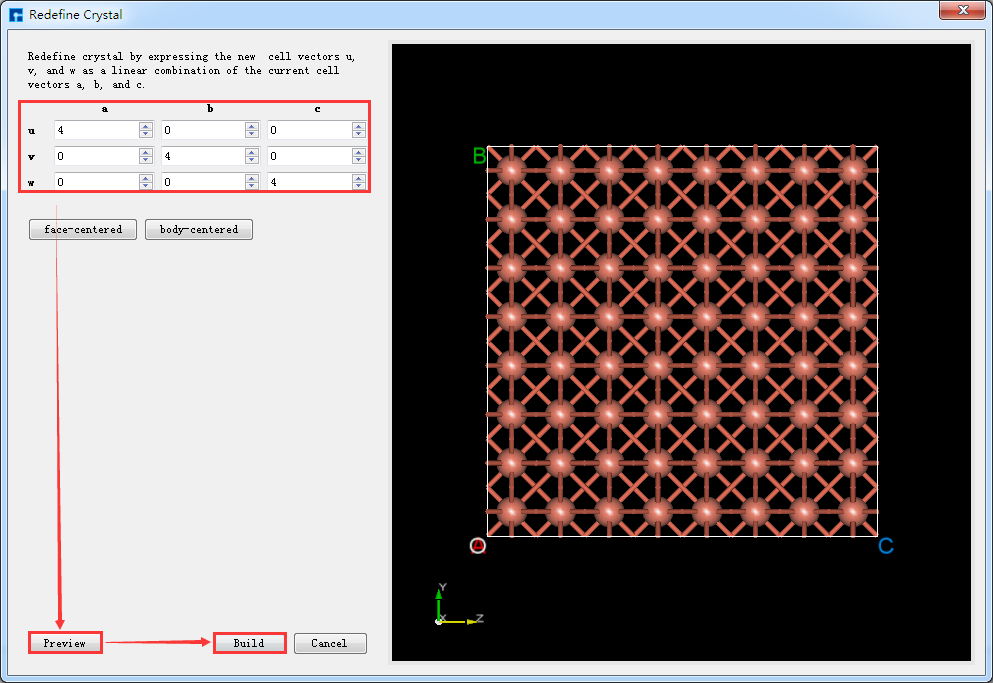
图5.6-5: 将Cu单胞扩为4*4*4超胞的操作界面¶
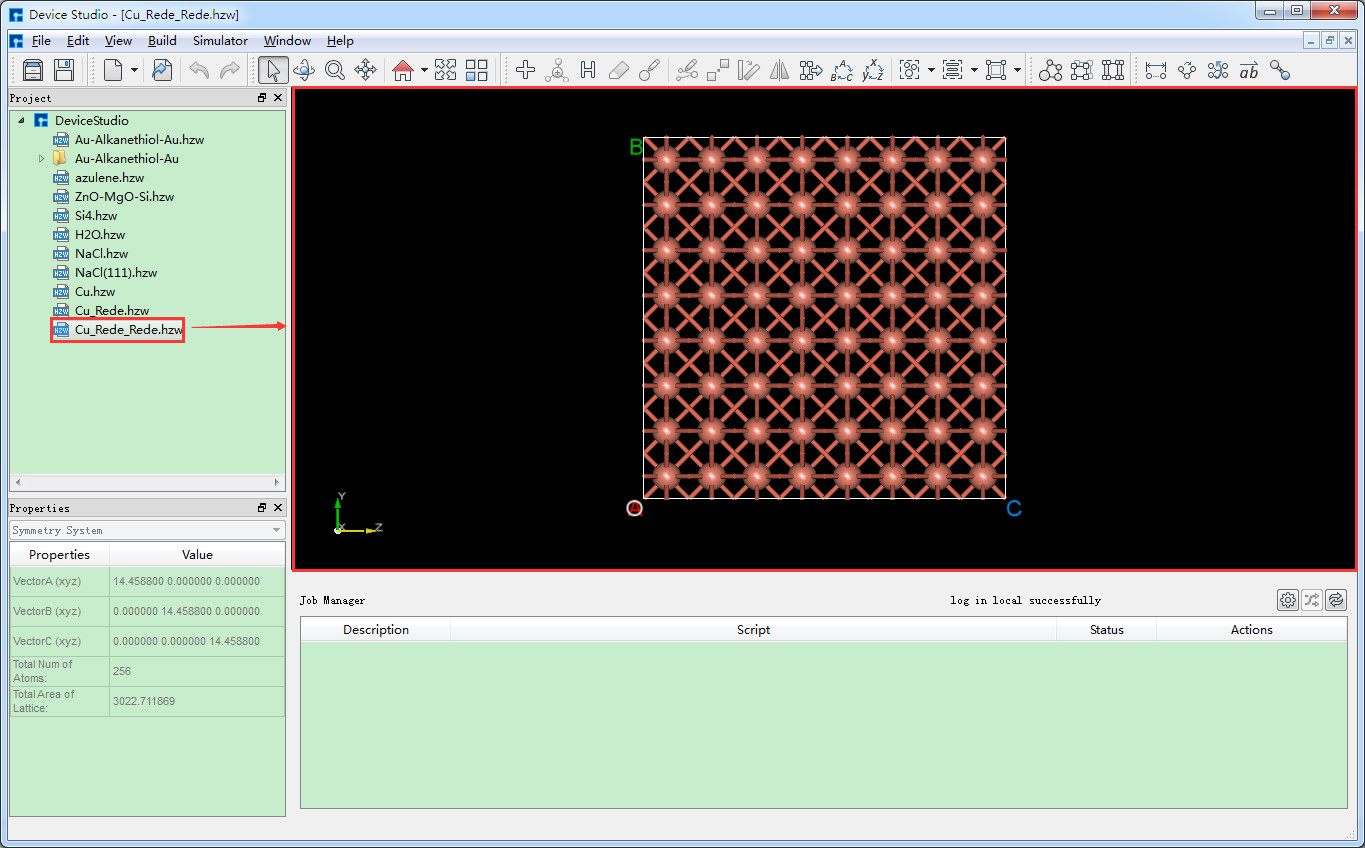
图5.6-6: Cu的超胞( Cu_Rede_Rede.hzw )结构在Device Studio中的3D显示¶
5.7. 器件建模¶
以构建金-烷硫醇-金(Au-Alkanethiol-Au)分子器件结构为例,以下将分为几个步骤逐步搭建并详细说明。
5.7.1. 导入Au晶体结构¶
从Device Studio本地数据库中导入Au晶体结构 ,即导入Au原胞,导入过程这里不做详细描述,用户可参考 本地数据库导入结构,导入Au晶体结构后的界面如图5.7-1所示。
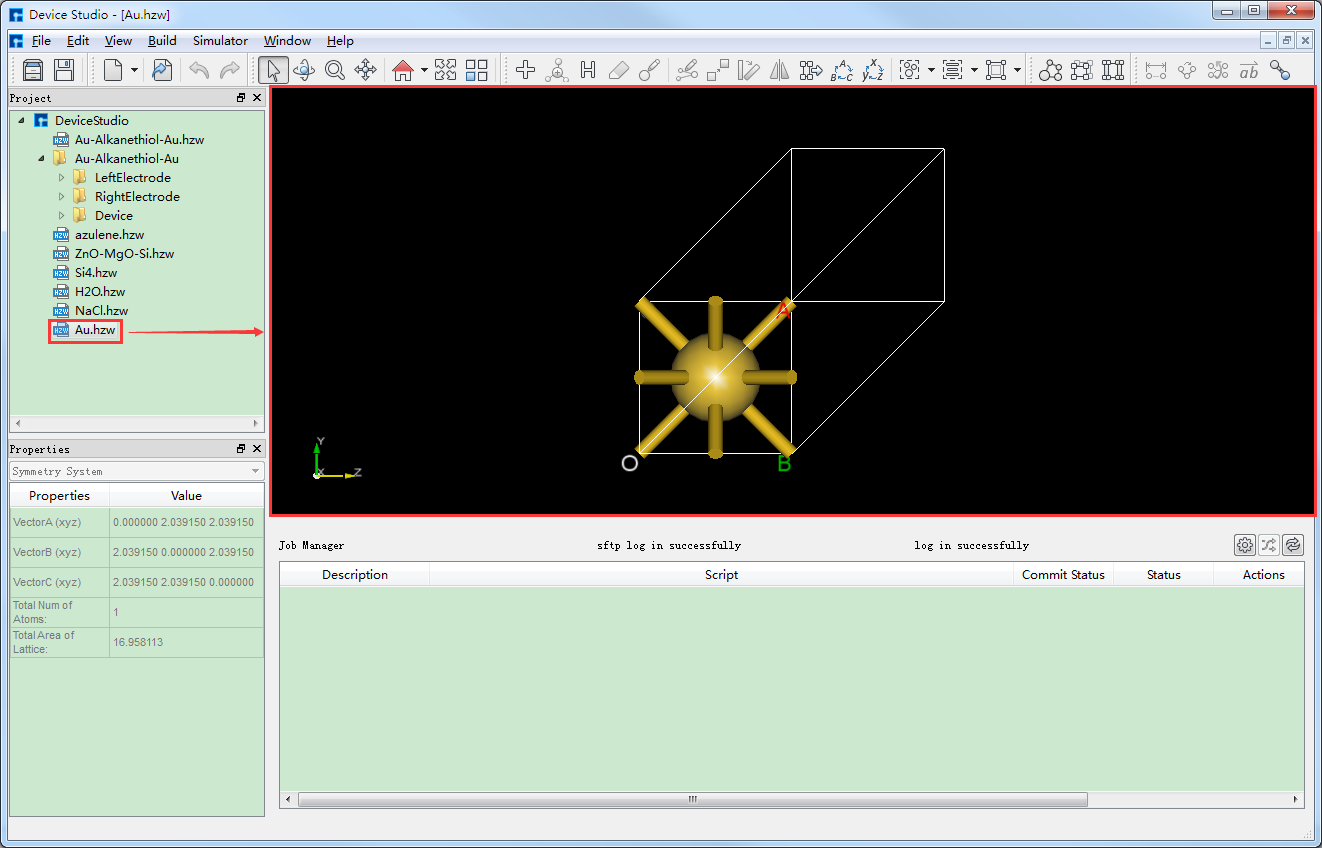
图5.7-1: 导入Au晶体结构后的界面¶
5.7.2. 将Au原胞转换为Au晶胞¶
点击图5.7-1中的 Build → Redefine Crystal 弹出Redefine Crystal界面如图5.7-2所示。点击图5.7-2中的 face-centered → Preview 则界面变化为如图5.7-3所示,用户可在图5.7-3的右侧区域预览转换后的结构,之后点击 Build 则Au原胞转换为Au晶胞结构,其结构文件保存在软件的项目管理区域(Project Explorer),同时可在3D显示区域查看到该结构的3D视图显示如图5.7-4所示。图5.7-4中的 Au.hzw 对应Au原胞,Au_Rede.hzw 对应Au晶胞,用户可根据计算需要选中结构文件,右击选择 Rename 对结构文件重新命名。
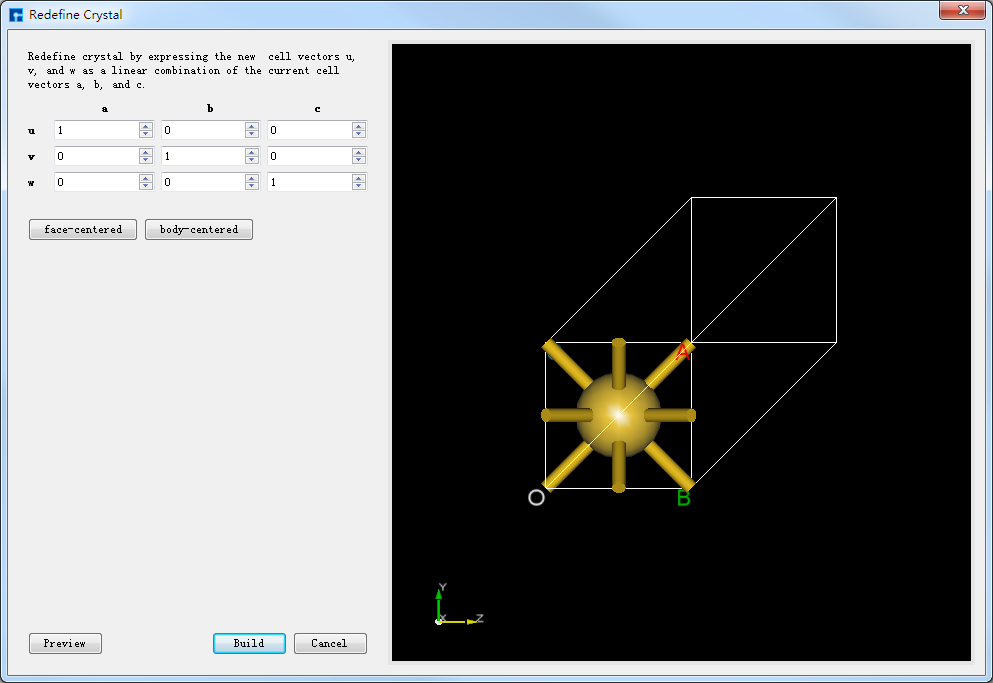
图5.7-2: 将Au原胞Redefine Crystal的界面¶
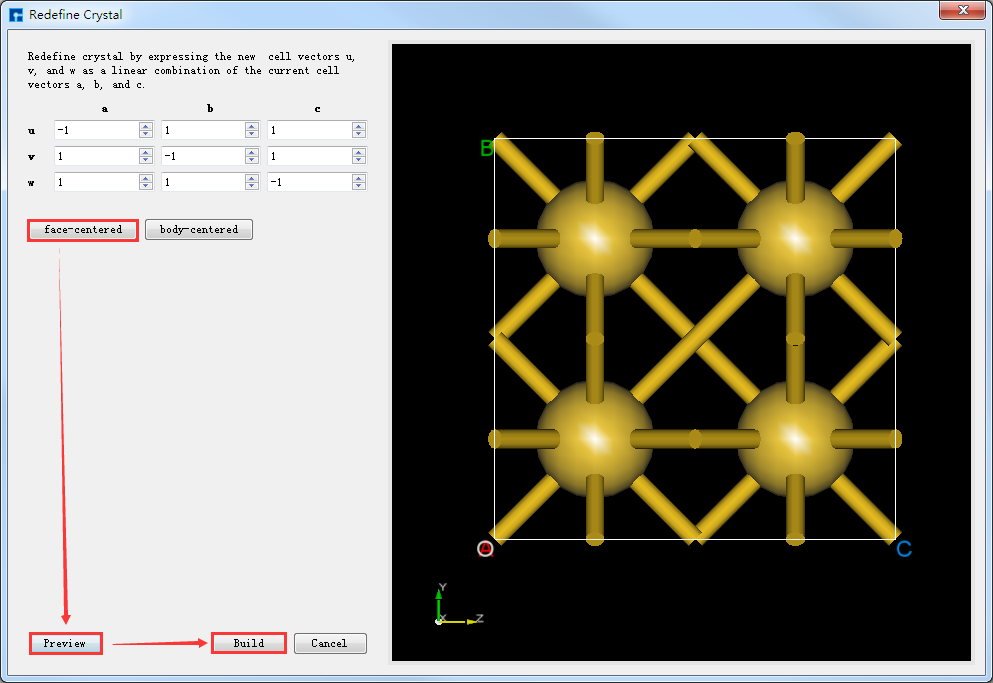
图5.7-3: 将Au原胞转换为Au晶胞的操作界面¶
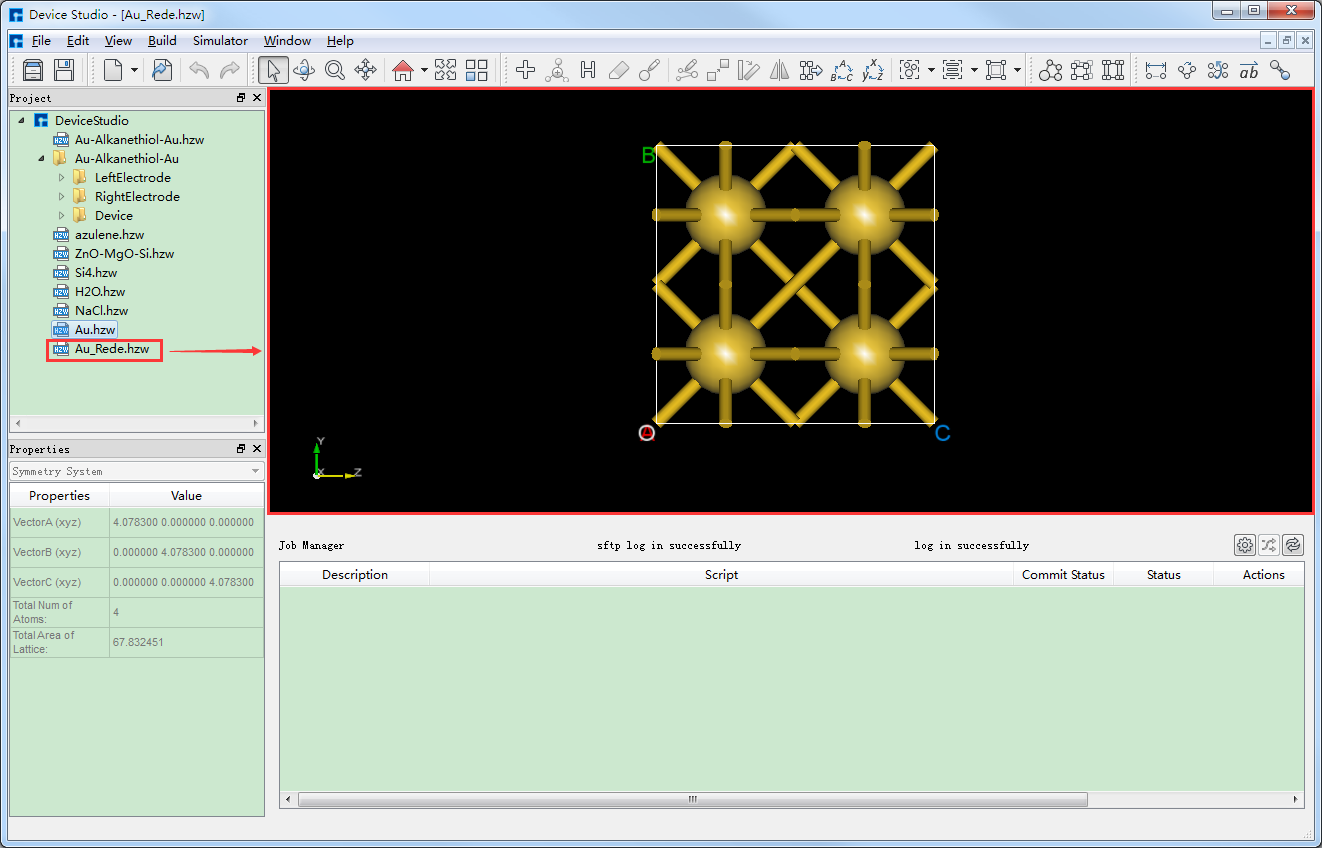
图5.7-4: 将Au原胞转换为Au晶胞后的界面¶
5.7.3. 将Au晶胞转换为Au超胞¶
将Au晶胞转换为2*2*4的超胞(简称:Au超胞),点击图5.7-4中的 Build → Redefine Crystal 弹出Redefine Crystal界面,按照如图5.7-5所示界面红色框选部分修改参数,点击 Preview 预览转换后的超胞结构,点击 Build 则Au晶胞转换为Au超胞结构,其结构文件保存在软件的项目管理区域(Project Explorer),同时可在3D显示区域查看到该结构的3D视图显示如图5.7-6所示。图5.7-6中的 Au_Rede_Rede.hzw 对应Au超胞,用户可根据计算需要给该结构文件重新命名。
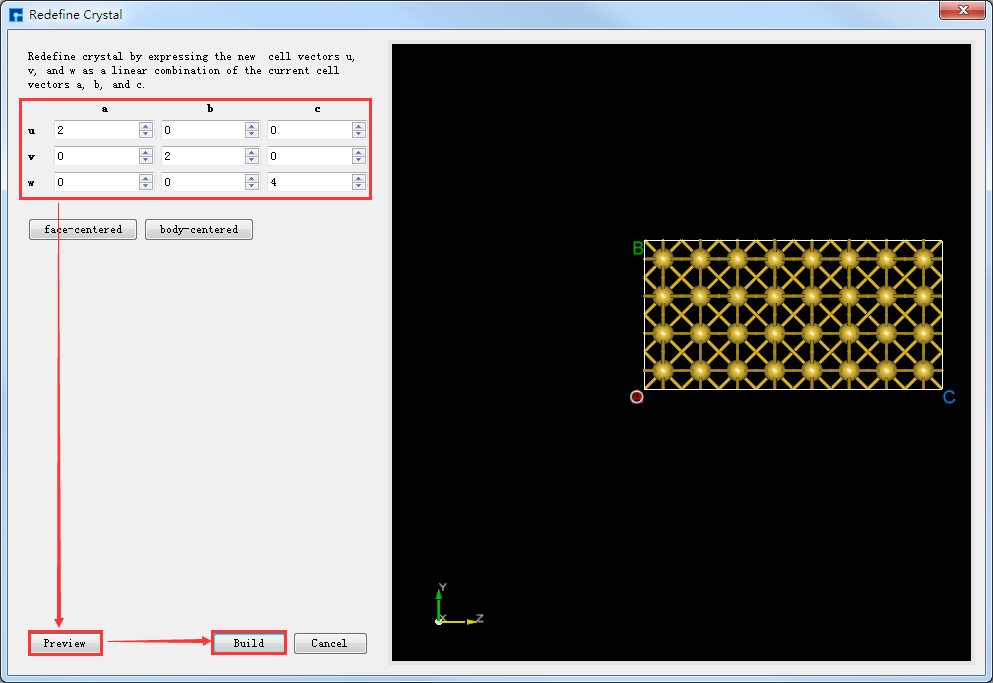
图5.7-5: 将Au晶胞转换为Au超胞的操作界面¶
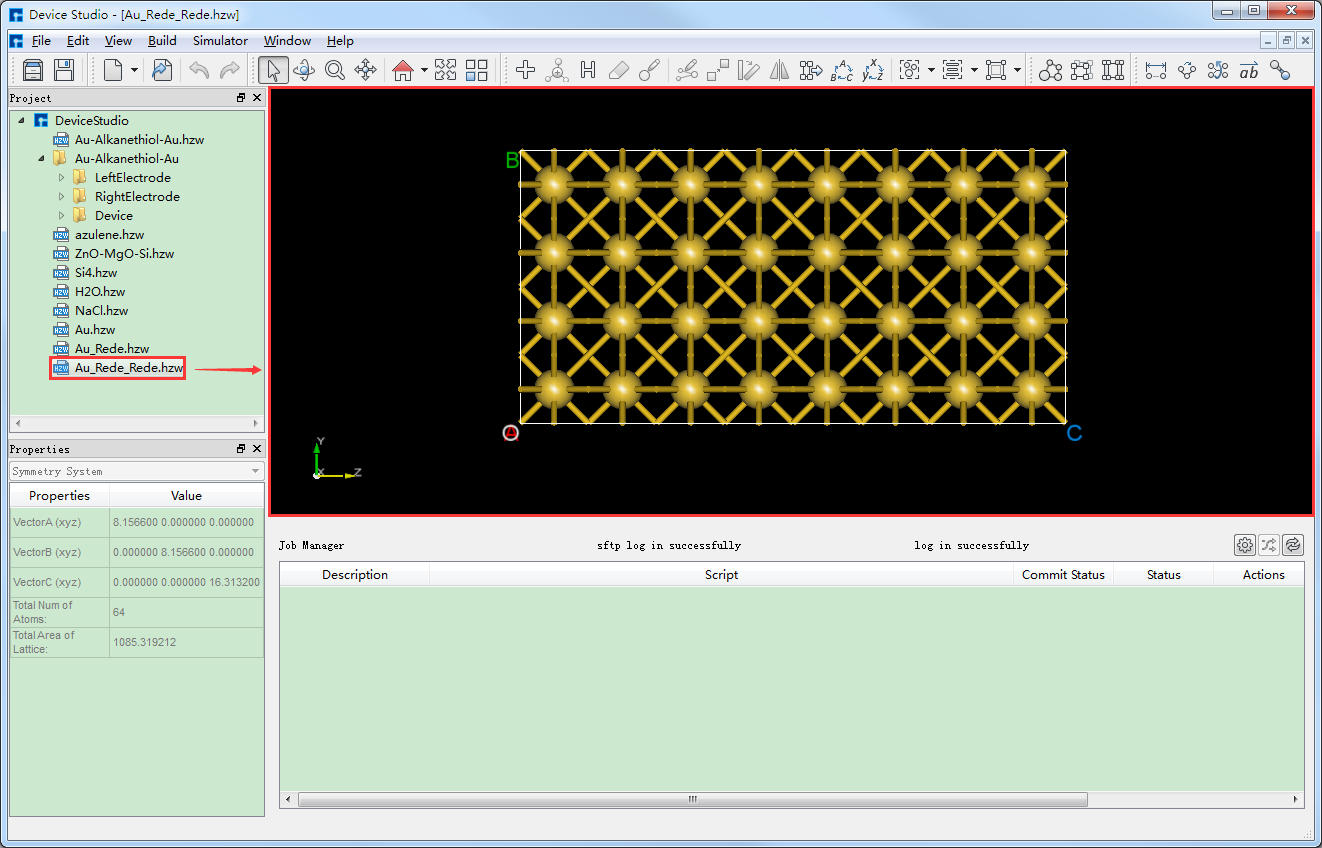
图5.7-6: 将Au晶胞转换为Au超胞后的界面¶
图5.7-7: CopyFile界面¶
5.7.4. 删除Au超胞中多余原子及操作¶
删除Au超胞中多余原子,并将结构在晶格单元中居中对齐。为避免删错原子无法还原,建议将Au超胞复制一份,选中 Au_Rede_Rede.hzw 结构文件 → 右击 → Copy,弹出CopyFile界面如图5.7-7所示,用户可根据计算需要重新命名,如Au_superCell,或直接采用默认命名,之后点击CopyFile对话框中的 OK 按钮则 Au_SuperCell.hzw 结构文件存储在项目管理区域,双击该结构文件,使之在3D视图显示如图5.7-8(a)所示。
(1)在3D显示区域,zy面情况下,用鼠标框选图5.7-8(a)中红色框部分,点击Toolbars上的 Delete Atom 快捷图标或点击键盘中 Delete 键删除选中的原子,删除后如图5.7-8(b)所示。
(2)点击Toolbars上的 3D Viewer zy View 快捷图标右侧的下拉按钮,选中 xy View 则由zy面切换为xy面如图5.7-8(c)所示,用鼠标框选图5.7-8(c)中红色框部分,点击Toolbars上的 Delete Atom 快捷图标或点击键盘 Delete 键删除选中的原子。删除后,点击Toolbars上的 3D Viewer zy View 快捷图标切换回zy面如图5.7-8(d)所示。 首先,鼠标框选图5.7-8(d)中的红色框部分;其次,按住键盘 Ctrl 键,同时鼠标点击图5.7-8(d)中的绿色框部分;最后,点击Toolbars上的 Delete Atom 快捷图标或点击键盘中 Delete 键删除选中的原子,删除后如图5.7-8(e)所示。
(3)在图5.7-8(e)的情况下,点击Toolbars上的 Center 快捷图标则可将结构中所有原子作为整体在晶格单元中居中对齐,居中对齐后如图5.7-8(f)所示。做完上述一些列操作,此时结构文件 Au_SuperCell.hzw 即Au_SuperCell超胞,对应着图5.7-8(f)的3D视图显示。
备注
由图5.7-8(a)可知被选中的原子为高亮黄色,用户可根据这点判断原子是否被选中。
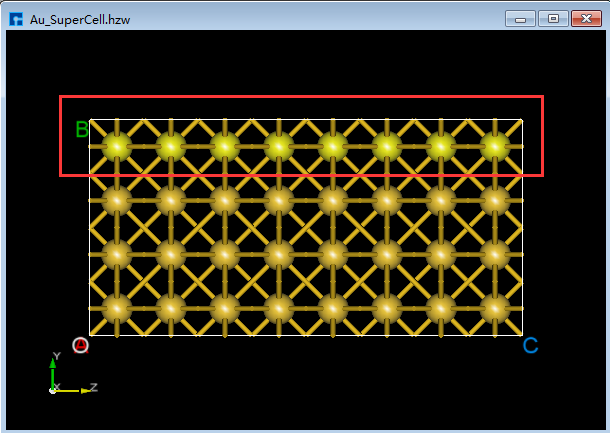
图5.7-8(a)¶ |
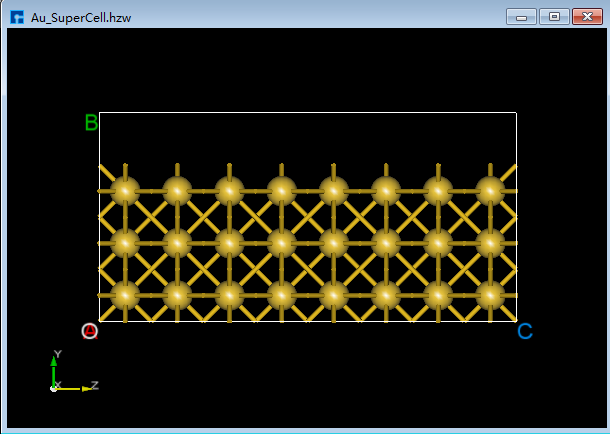
图5.7-8(b)¶ |
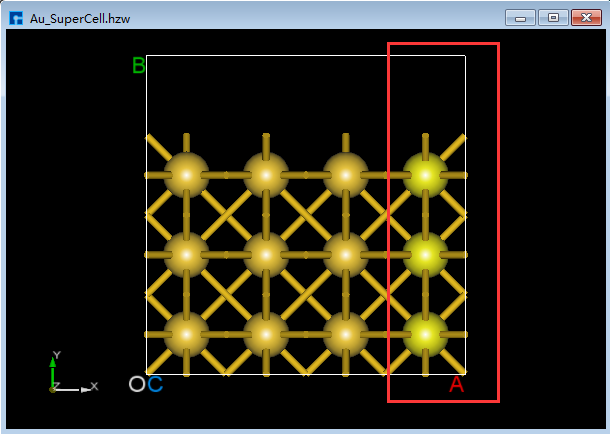
图5.7-8(c)¶ |
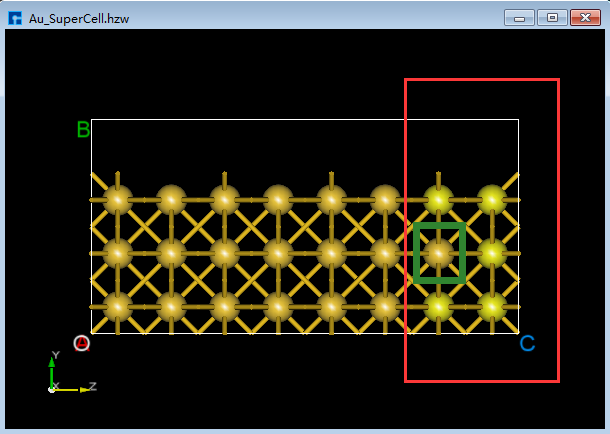
图5.7-8(d)¶ |
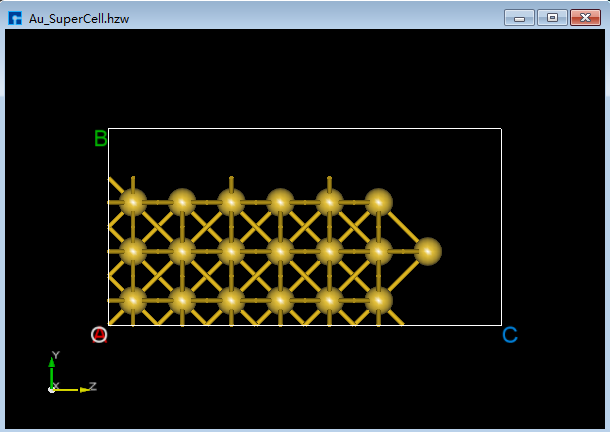
图5.7-8(e)¶ |
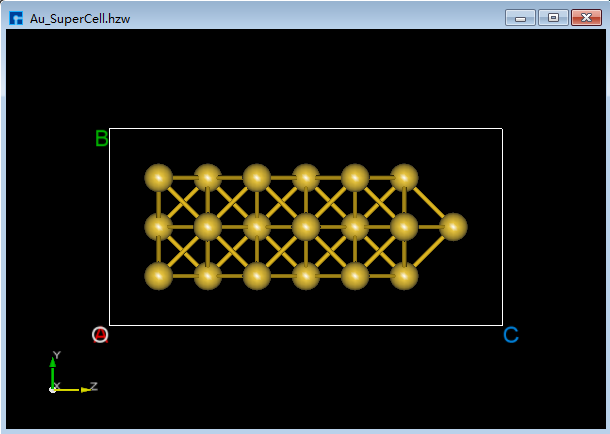
图5.7-8(f)¶ |
5.7.5. 设置X和Y轴方向为真空¶
将Au_SuperCell超胞的X和Y轴方向设置为真空。将Au_SuperCell超胞复制一份并重命名为Au_SuperCell_1,双击该结构文件 Au_SuperCell_1.hzw 使之在3D显示区域显示。点击Toolbars上的 Convert to Crystal 快捷图标弹出Convert to Crystal界面,按照图5.7-9所示界面红色框选部分设置参数 → 界面右侧预览 → Build 则将Au_SuperCell超胞的X和Y轴方向设置为真空如图5.7-10(a)所示,点击Toolbars上的 Center 快捷图标则可将结构中所有原子作为整体在晶格单元中居中对齐,居中对齐后如图5.7-10(b)所示。做完上述一些列操作,结构文件 Au_SuperCell_1.hzw 即Au_SuperCell_1超胞,对应着图5.7-10(b)的3D视图显示。
备注
将结构文件复制并重命名,是为了在教程中方便说明,同时避免一系列操作后失误后无法还原,用户可根据建模需要决定是否复制、重命名。一般误删或误操作,可点击Toolbars上的 Undo 快捷图标撤销,或按快捷键 Ctrl+Z 撤销。
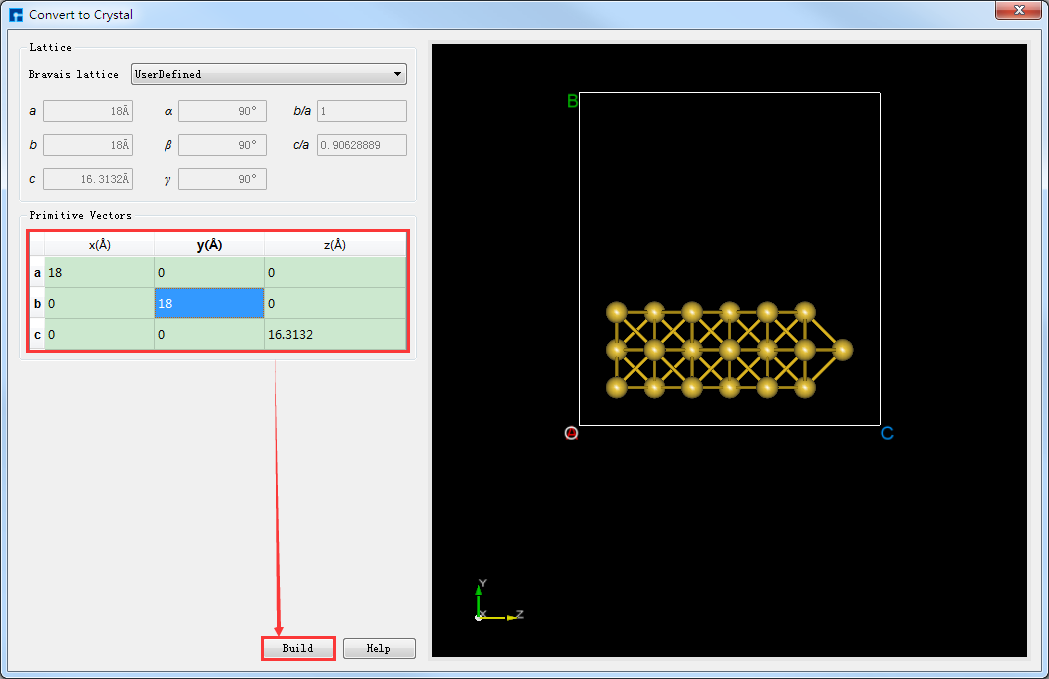
图5.7-9: Convert to Crystal界面¶
备注
在图5.7-9界面中点击 Build 按钮后 Au_SuperCell.hzw 结构的3D视图不显示在3D显示区域的合适位置,用户可通过点击Toolbars上 3D Viewer zy View 快捷图标或按快捷键 Ctrl+R 重置该结构的3D视图到合适位置,重置后的结构3D视图如图5.7-10(b)所示。用户在建模过程中遇到结构的3D视图不在3D显示区域的合适位置,均可通过上述方式重置。
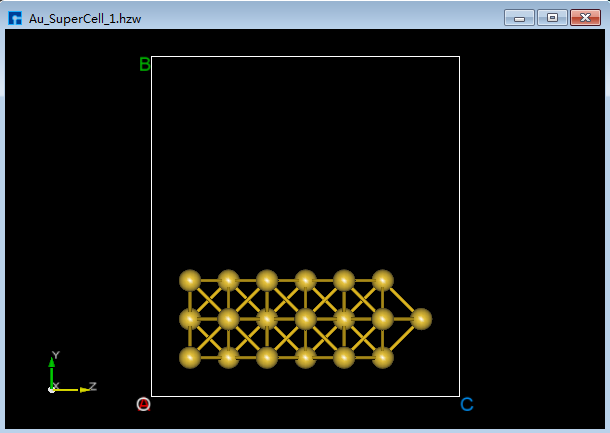
图5.7-10(a)¶ |
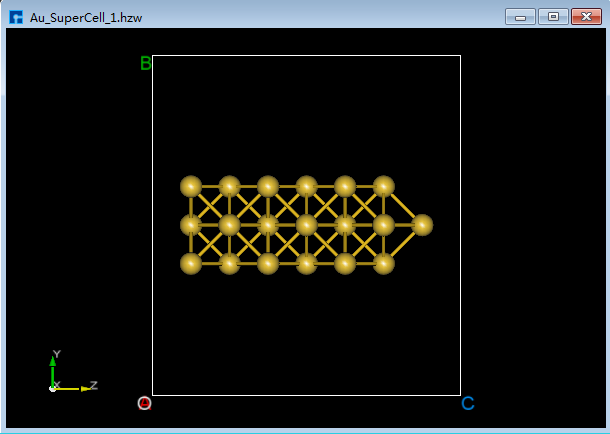
图5.7-10(b)¶ |
5.7.6. 将结构做镜像处理并重置晶格常数¶
将选中的原子做镜像处理,重新设置晶格常数,并将结构居中对齐。如图5.7-11所示,选中所有原子 → 点击 Mirror Atom 快捷图标 → 弹出Mirror界面 → 设置参数 → 勾选Copy → 点击 Apply,则结构如图5.7-13(a)所示。点击 Convert to Crystal 快捷图标弹出Convert to Crystal界面如图5.7-12所示,按图中红色框选部分设置参数后,在界面右侧预览重置晶格常数后的结构,点击 Build,点击 Center 快捷图标将重置晶格常数后的结构中所有原子作为整体在晶格单元中居中对齐,对齐后如图5.7-13(b)所示。
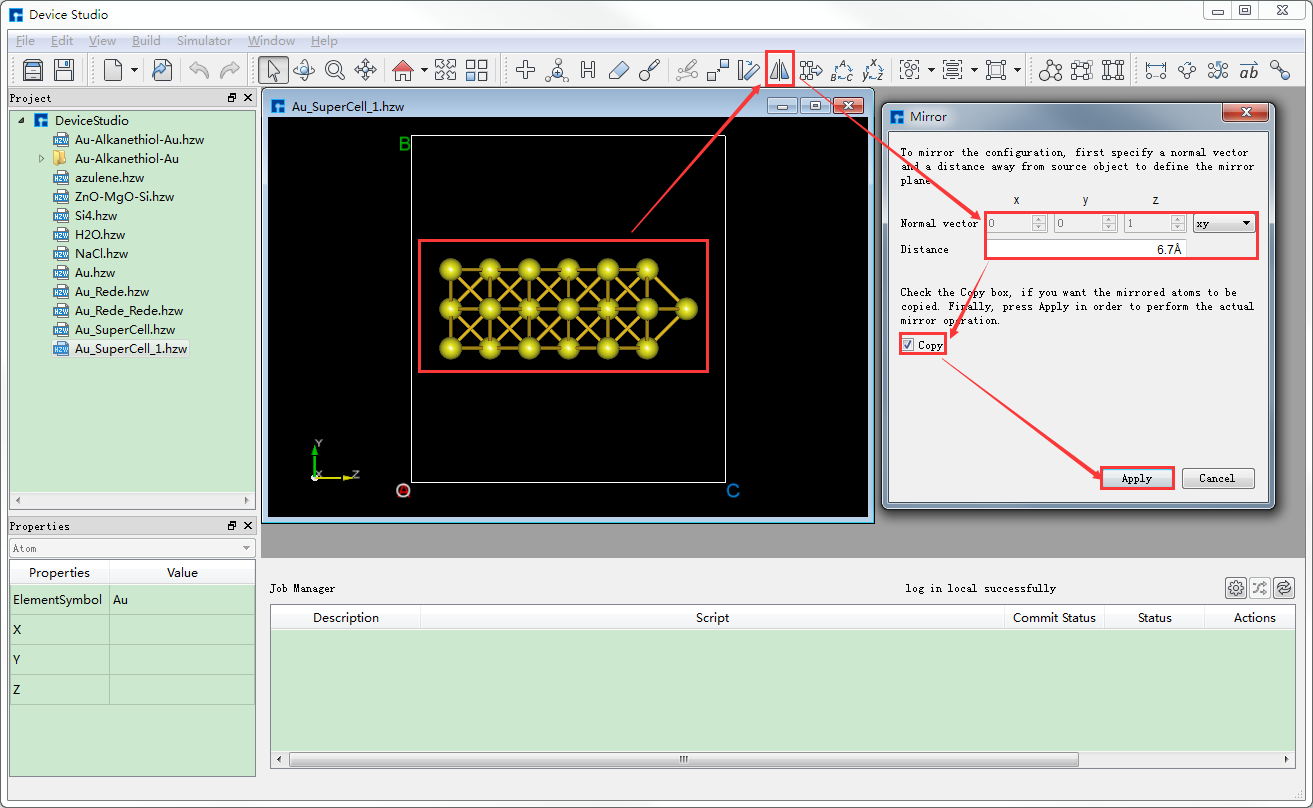
图5.7-11: 对Au_SuperCell_1超胞做镜像处理操作界面¶
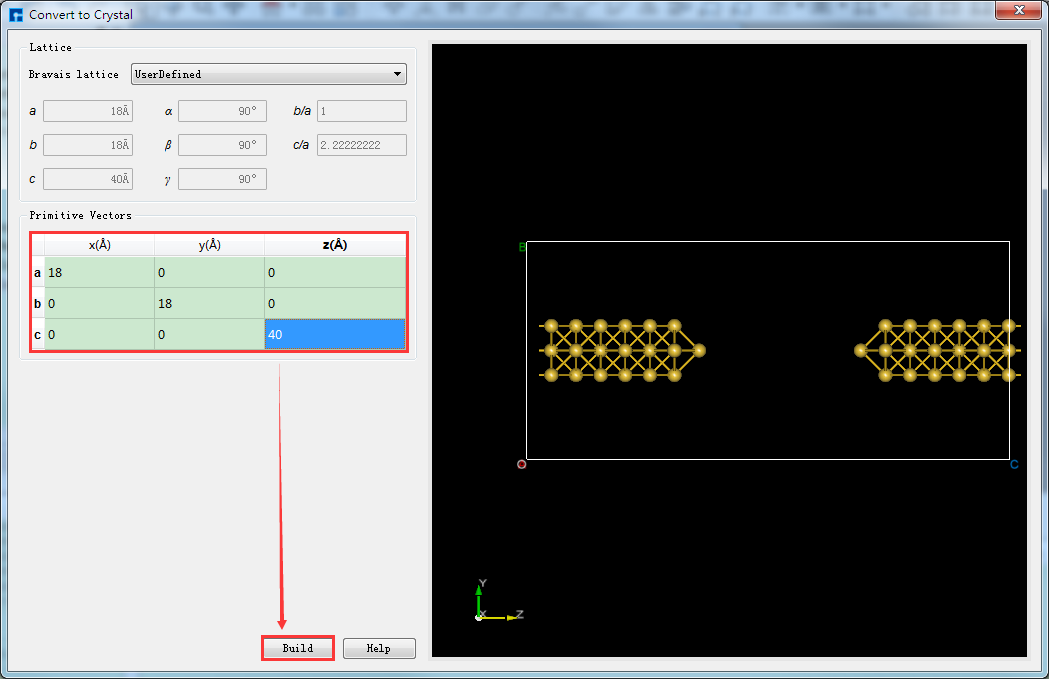
图5.7-12: 对Au_SuperCell_1超胞重新设置晶格常数操作界面¶
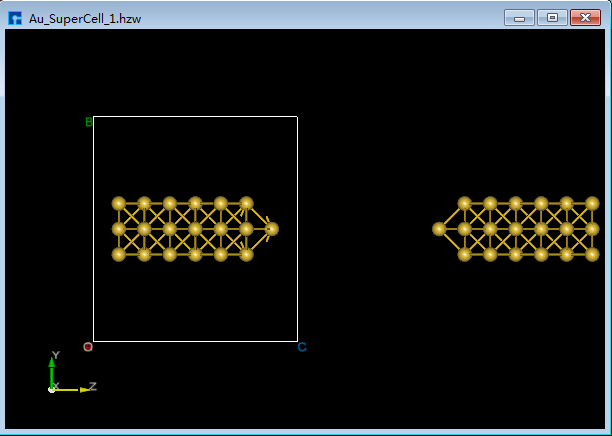
图5.7-13(a)¶ |
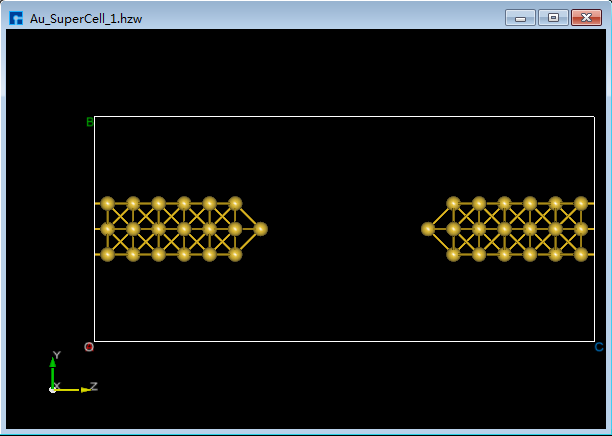
图5.7-13(b)¶ |
5.7.7. 构建两端口器件结构¶
导入烷硫醇分子结构,并将结构复制到Au_SuperCell_1超胞结构中居中位置,之后构建两端口器件结构。
(1)导入烷硫醇(Alkanethiol)分子结构如图5.7-14所示。
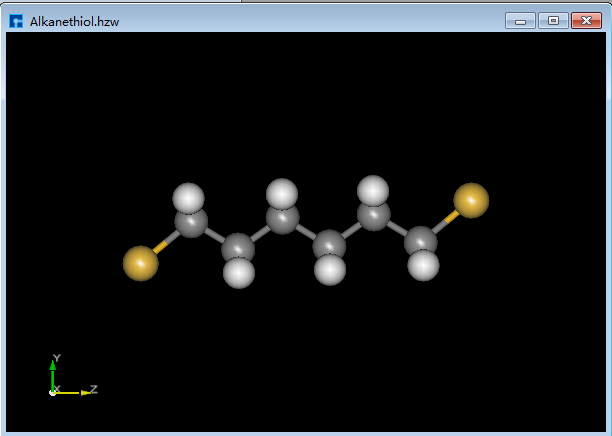
图5.7-14: 烷硫醇(Alkanethiol)分子结构¶
(2)双击 Au_SuperCell_1.hzw 打开Au_SuperCell_1超胞结构,鼠标框选并按快捷键 Ctrl+C 复制烷硫醇(Alkanethiol)分子结构;鼠标点击图5.7-13(b)界面左侧位置,并按快捷键 Ctrl+V 将烷硫醇(Alkanethiol)分子结构粘贴到Au_SuperCell_1超胞结构中如图5.7-15(a)所示。为方便将粘贴的烷硫醇(Alkanethiol)分子结构移动到Au_SuperCell_1超胞结构的居中位置,点击 3D Viewer zy View 快捷图标右侧的下拉按钮,选中 zx View 则由zy面切换为zx面如图5.7-15(b)所示。点击快捷图标 Center 则将烷硫醇(Alkanethiol)分子结构移动到Au_SuperCell_1超胞结构的居中位置如图5.7-15(c)所示,点击快捷图标 3D Viewer zy View 则切换为zy面如图5.7-15(d)所示。
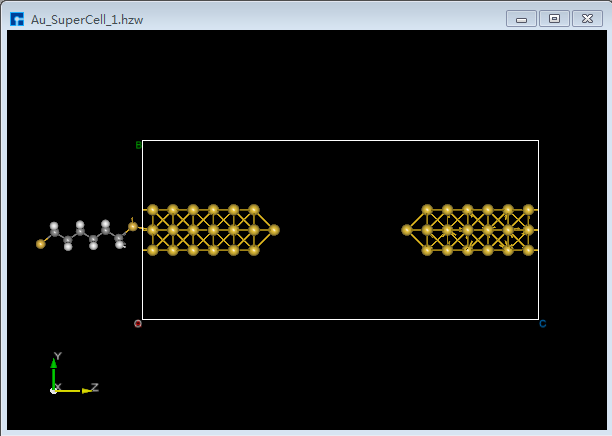
图5.7-15(a)¶ |
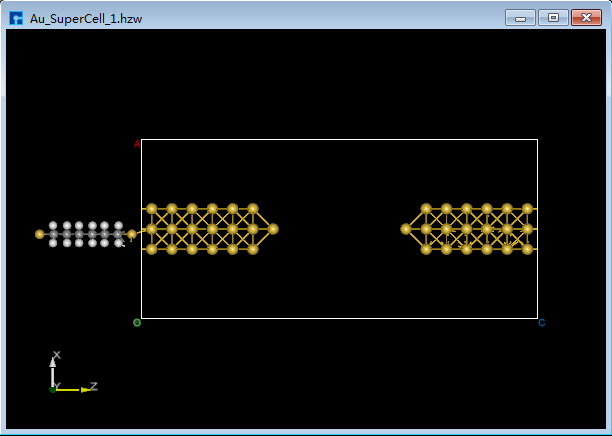
图5.7-15(b)¶ |
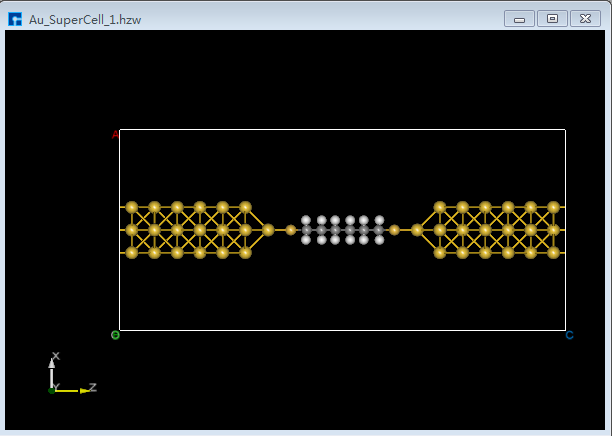
图5.7-15(c)¶ |
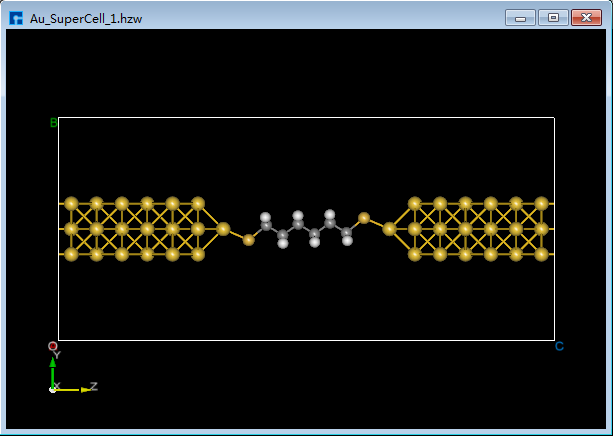
图5.7-15(d)¶ |
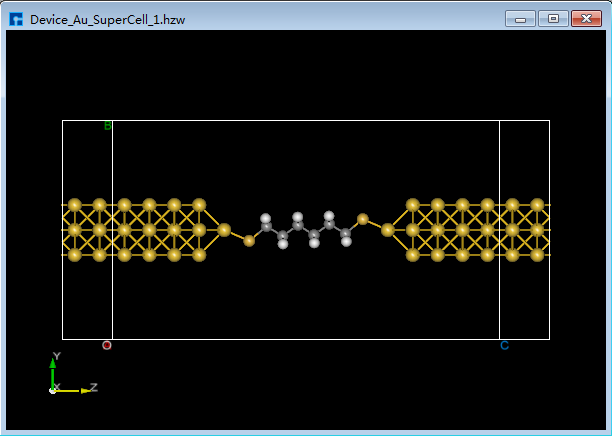
图5.7-15(e)¶ |
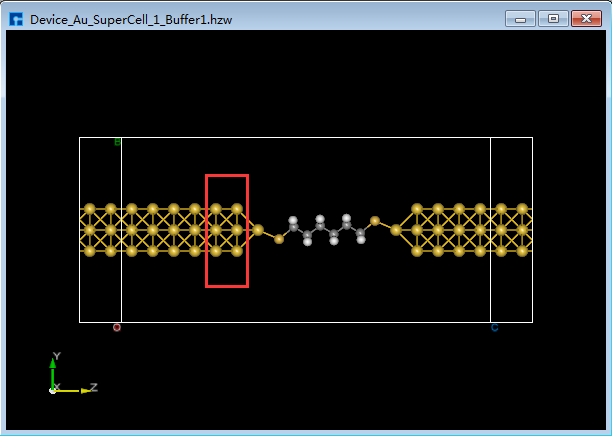
图5.7-15(f)¶ |
(3)点击 Convert to Device 快捷图标,弹出弹出Convert to Device界面如图5.7-16所示,分别勾选界面中的红色框部分,点击 Preview 在界面右侧预览构建的两端口器件结构,点击 Build 则搭建好的两端口器件结构 Device_Au_SuperCell_1.hzw 如图5.7-15(e)所示。
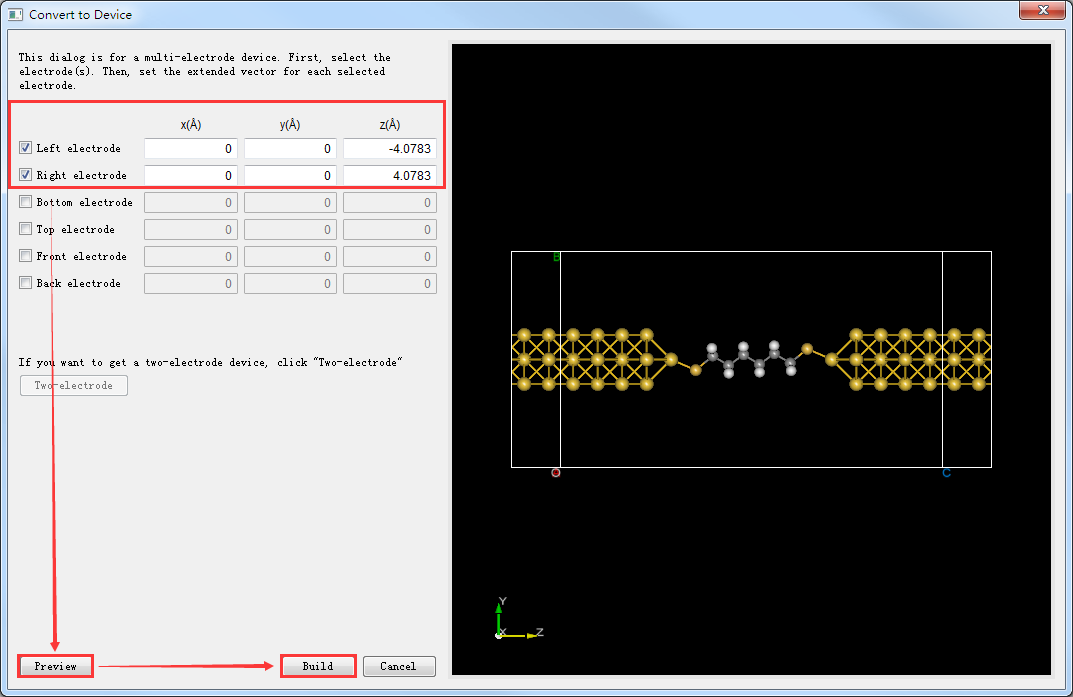
图5.7-16: Convert to Device界面¶
5.7.8. 给器件结构添加缓冲层¶
用户可根据计算需要决定是否给构建好的两端口器件结构增加缓冲层,无强制要求。以沿着输运方向(Z轴方向),给器件结构 Device_Au_SuperCell_1.hzw 靠近电极添加缓冲层为例,双击打开该器件结构 → 点击软件Menu菜单栏的 Simulator → Nanodcal → Add Buffer,弹出Add Buffer界面如图5.7-17所示,点击界面中Left后的 + 号按钮2次,可在界面右侧预览到沿着输运方向器件的左侧添加了缓冲层,点击 Build 则添加了缓冲层的两端口器件结构 Device_Au_SuperCell_1_Buffer1.hzw 如图5.7-15(f)所示。
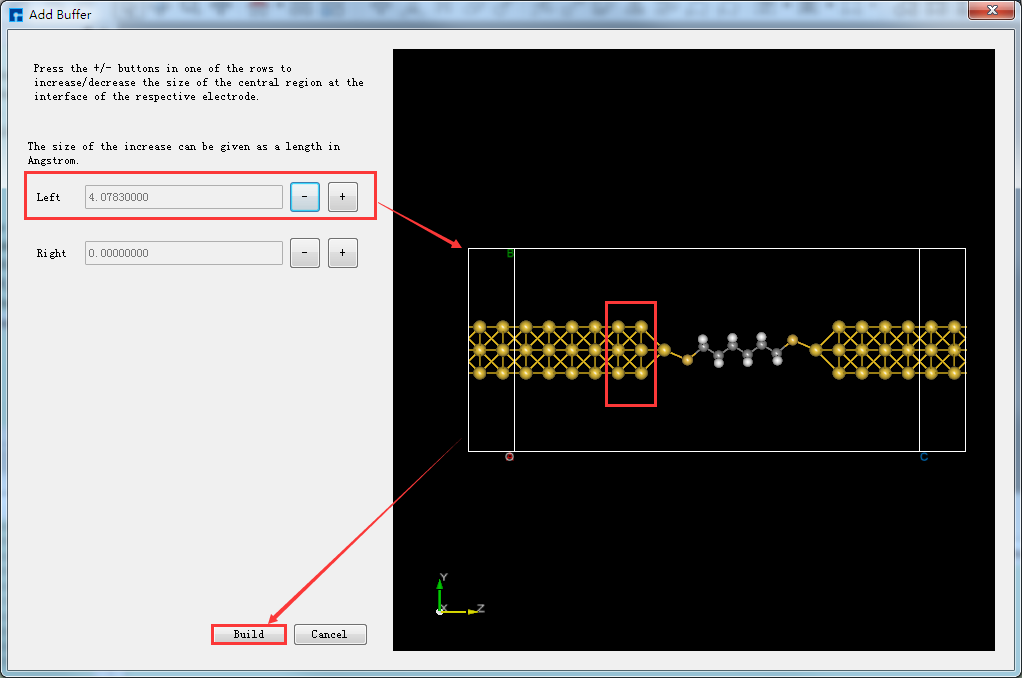
图5.7-17: Add Buffer界面¶
备注
上述给器件结构添加缓冲层只是案例,用户可根据计算需要决定是否在靠近电极两端添加缓冲层,决定添加几层。
5.8. 导出结构¶
对于搭建好的结构,如两端口器件结构 Device_Au_SuperCell_1_Buffer1.hzw ,若想导出此结构文件,双击打开该结构文件 → 点击软件的 File → Export 弹出导出当前结构文件界面,用户可根据需要选择存储位置,对该结构文件命名,或采用默认命名,并选择存储格式。
6. 结构操作¶
对于结构的操作,用户可先仔细阅读 工具栏(Toolbars) 中各快捷图标的功能描述,再根据需要对结构进行相应操作,操作后的结构视图均可在3D显示区域查看。
6.1. 结构的3D显示基础操作¶
Device Studio支持从ZY、XY、XZ、YZ、YX和ZX面观察结构的3D视图,如在3D显示区域的NaCl(1 1 1)晶体结构,用户通过鼠标点击工具栏中的 3D Viewer zy View 快捷图标将结构重置到ZY面,通过点击该快捷图标的下拉按钮可选择从XY、XZ、YZ、YX或ZX面来观察NaCl(1 1 1)晶体结构的3D视图,NaCl(1 1 1)晶体结构在3D显示区域的ZY、XY、XZ、YZ、YX和ZX面的3D视图分别为6.1-1(a)、6.1-1(b)、6.1-1(c)、6.1-1(d)、6.1-1(e)和6.1-1(f)所示。
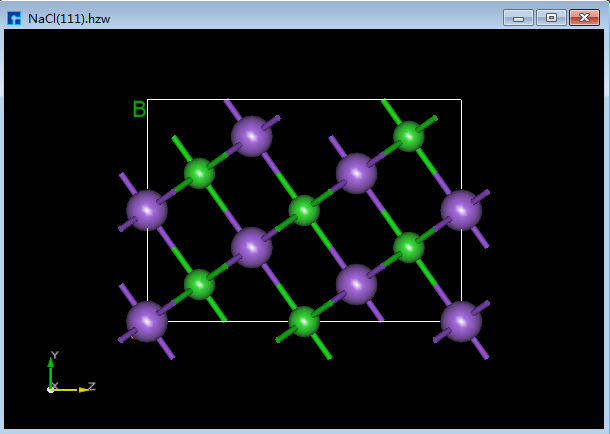
图6.1-1(a): NaCl(1 1 1)ZY面3D视图¶ |
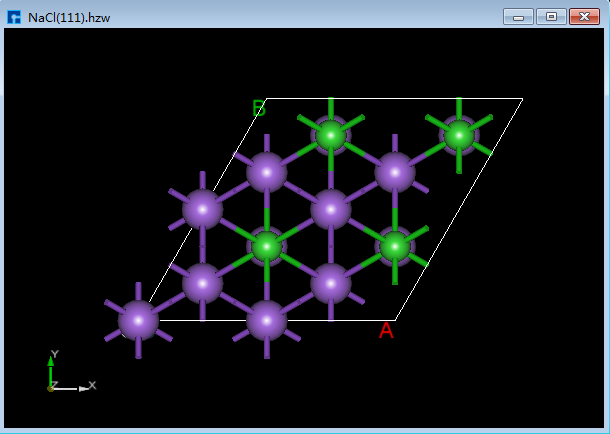
图6.1-1(b): NaCl(1 1 1)XY面3D视图¶ |
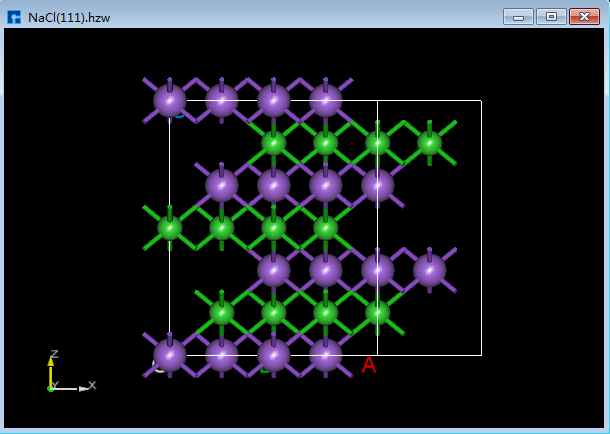
图6.1-1(c): NaCl(1 1 1)XZ面3D视图¶ |
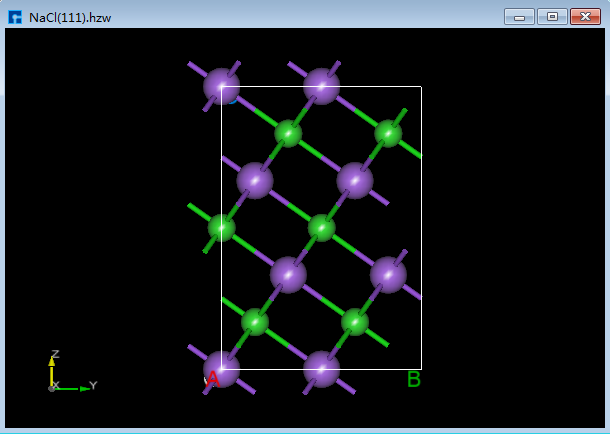
图6.1-1(d): NaCl(1 1 1)YZ面3D视图¶ |
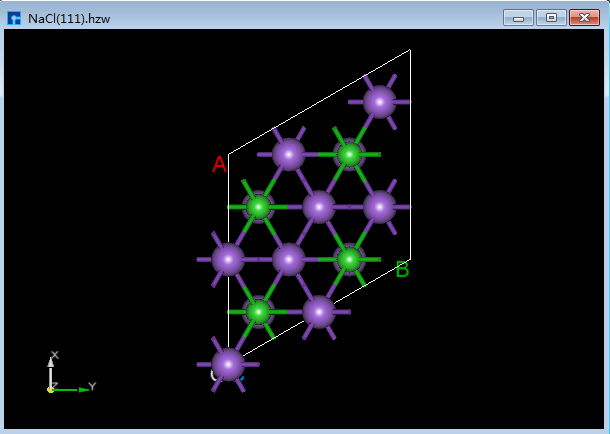
图6.1-1(e): NaCl(1 1 1)YX面3D视图¶ |
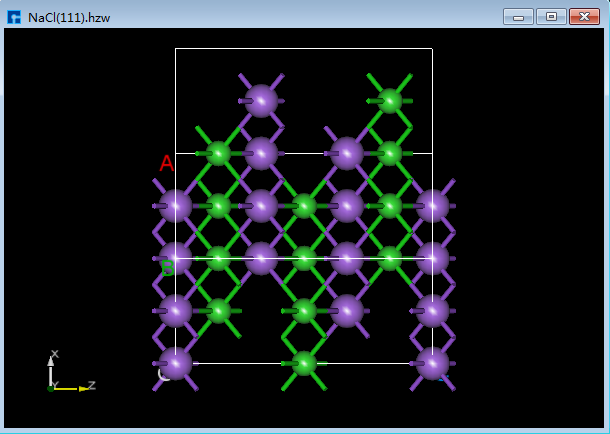
图6.1-1(f): NaCl(1 1 1)ZX面3D视图¶ |
在结构3D显示区域,用户可通过滚动鼠标中键将结构的3D视图放大或缩小;可先选中工具栏中的 3D Viewer Translation Mode 快捷图标或按住鼠标中键,通过拖动鼠标将结构的3D视图在该区域平移;可先选中工具栏中的 3D Viewer Rotation Mode 快捷图标或按住鼠标右键,通过拖动鼠标将结构的3D视图在该区域旋转。
6.2. 结构的修改操作¶
以晶体MoS 2 为例来说明结构的一系列修改操作,准备结构文件 MoS2.hzw,鼠标拖动该文件到软件的Project Explorer(项目管理)区域即可导入结构,结构的zy面3D视图如图6.2-1(a)所示。
6.2.1. 添加原子¶
如在晶体MoS 2 中添加一个Cr原子,具体操作为点击Toolbars上的 Add Atom 快捷图标则弹出元素周期表,在元素周期表中选中Cr,点击 OK,在图6.2-1(a)所示界面中根据需要在某个位置点击鼠标左键,再点击重新计算键长 Recalculate LinkerBond 快捷图标则完成在MoS 2 中添加一个Cr原子的操作,完成后的3D视图如图6.2-1(b)所示。
6.2.2. 删除原子¶
如删除如图6.2-1(b)所示的结构中左边的S原子,具体操作为选中想删除的S原子,点击Toolbars上的 Delete Atom 快捷图标或直接按键盘中 Delete 键则可删除S原子,之后点击重新计算键长 Recalculate LinkerBond 快捷图标,删除S原子后的3D视图如图6.2-1(c)所示。
6.2.3. 替换原子¶
如将如图6.2-1(c)所示的结构中的Cr原子替换成W原子,具体操作为选中想替换的Cr原子,点击Toolbars上的替换原子 Replace Atom 快捷图标则弹出元素周期表,在元素周期表中选中W,点击 OK,再点击重新计算键长 Recalculate LinkerBond 快捷图标,则完成将如图6.2-1(c)所示的结构中的Cr原子替换成W原子的操作,完成后的结构的3D视图如图6.2-1(d)所示。
6.2.4. 修改原子坐标¶
如将图6.2-1(d)中W原子的坐标位置由(0.5951619,1.47565329,7.18570423)修改为(1.0,1.5,12.0),具体操作为先鼠标选中W原子,则在Properties Explorer(结构属性区域)查看到选中的W原子的坐标信息为(0.5951619,1.47565329,7.18570423),再鼠标分别双击合适位置填写1.0、1.5和12.0,则修改好W原子的坐标,最后点击重新计算键长 Recalculate LinkerBond 快捷图标,修改原子坐标后结构的3D视图如图6.2-1(e)所示。
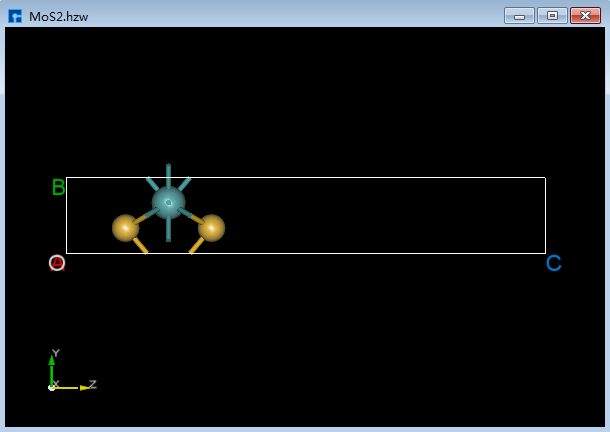
图6.2-1(a)¶ |
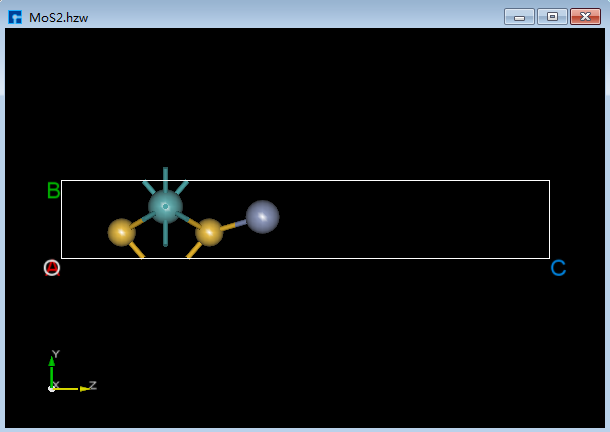
图6.2-1(b)¶ |
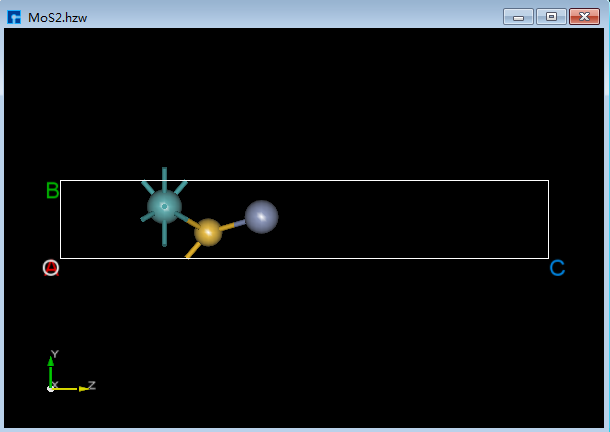
图6.2-1(c)¶ |
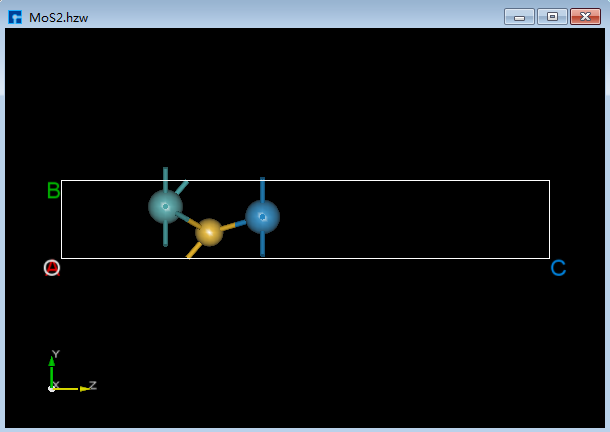
图6.2-1(d)¶ |
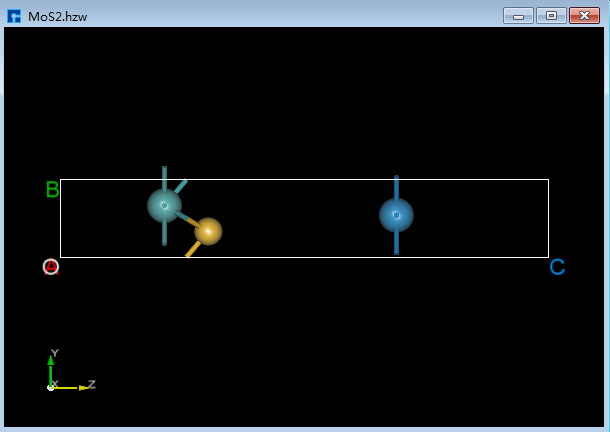
图6.2-1(e)¶ |
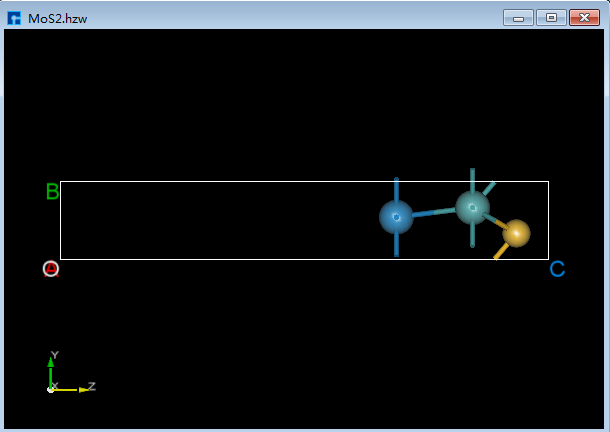
图6.2-1(f)¶ |
6.2.5. 移动选中的原子¶
对于图6.2-1(e)所示的结构,移动左边的两个原子,具体操作如图6.2-2红色部分所示,先鼠标框选左边两个原子,点击 Move Atom 快捷图标,则弹出Move Atom界面,在界面中填写移动距离,点击沿着Z的正方向移动,移动之后点击新计算键长 Recalculate LinkerBond 快捷图标,移动后的结构的3D视图如图6.2-1(f)所示。
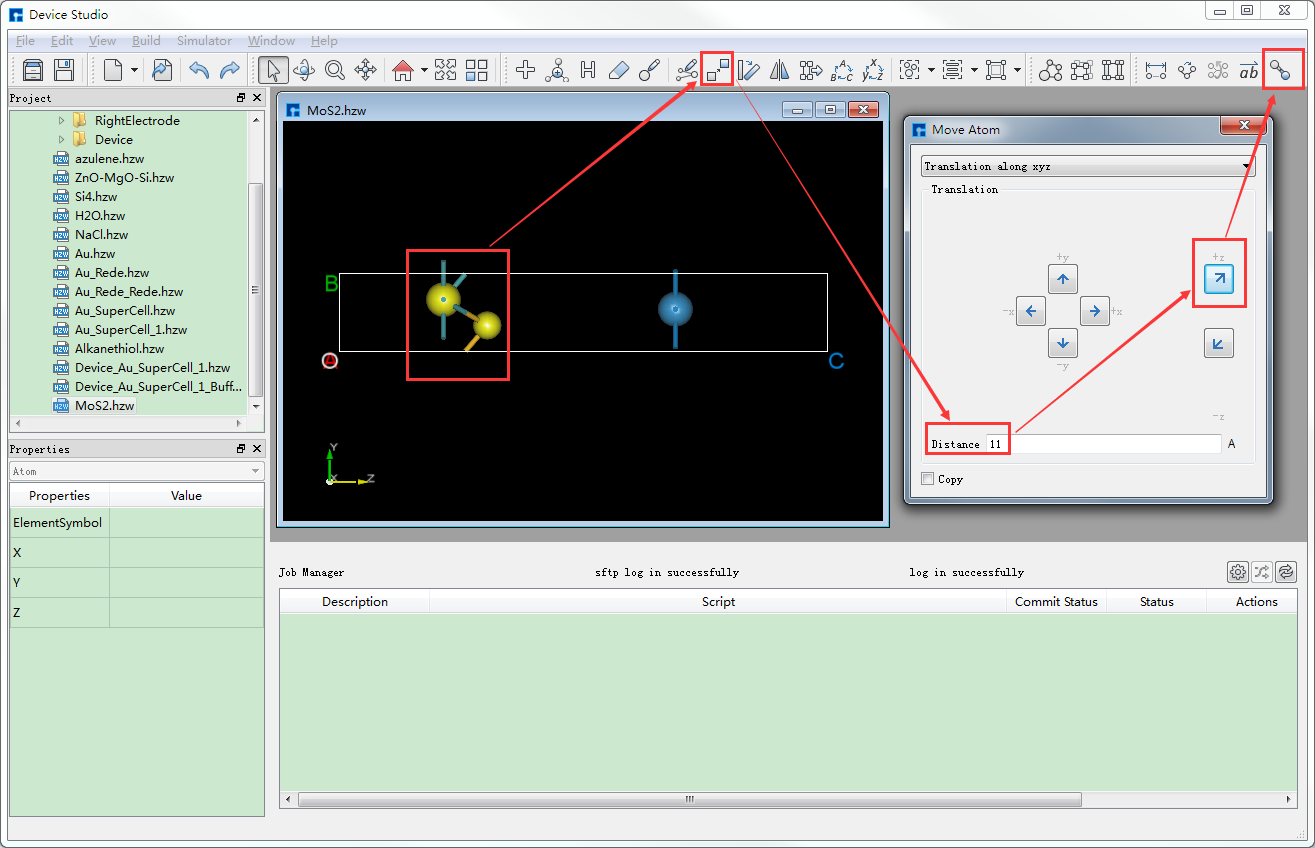
图6.2-2: 移动选中的原子操作界面¶
备注
这里对于结构的修改操作只描述了一部分,用户可根据需要仔细阅读 工具栏(Toolbars) 中各快捷图标的功能描述,再进行相应的结构操作。
6.3. 结构的信息测量¶
以Si8晶体结构为例描述结构的信息测量一系列操作。准备结构文件 Si8.hzw,鼠标拖动该文件到软件的Project Explorer(项目管理区域)即可导入结构,结构的zy面3D视图如图6.3-1所示。
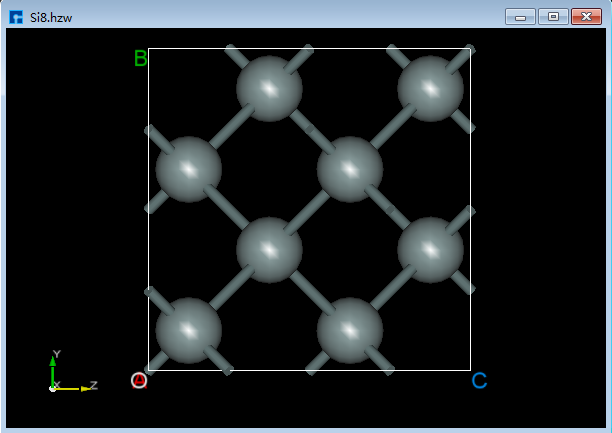
图6.3-1: Si8晶体结构¶
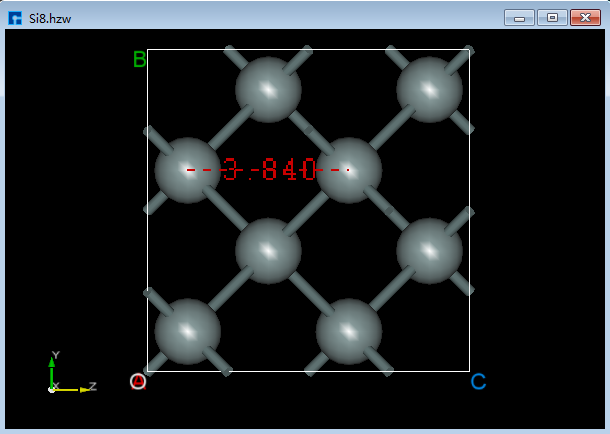
6.3.1. 测量2个原子之间的距离¶
对于图6.3-1所示的晶体结构,先点击Toolbars上的 Distance 快捷图标,再通过鼠标分别选中结构中的2个原子则可测量选中的2个原子之间的距离如图6.3-2(a)所示,若不想保留测量数字,点击 3D Viewer Selection Mode 快捷图标,选中测量数字,按键盘中的 Delete 即可。
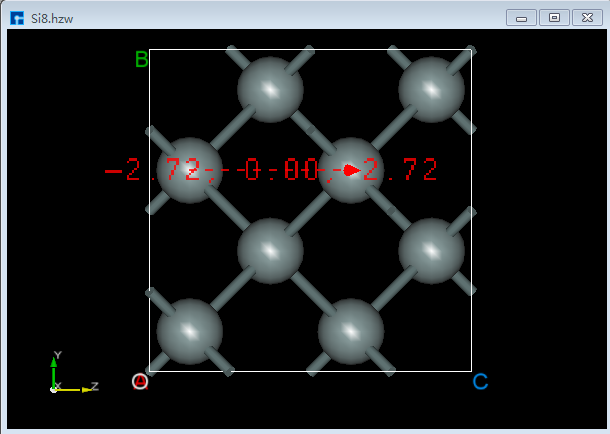
6.3.2. 测量2个原子之间的向量¶
对于图6.3-1所示的晶体结构,先点击Toolbars上的 Vector between two atoms 快捷图标,再通过鼠标分别选中结构中的2个原子则可测量选中的2个原子之间的向量如图6.3-2(b)所示。
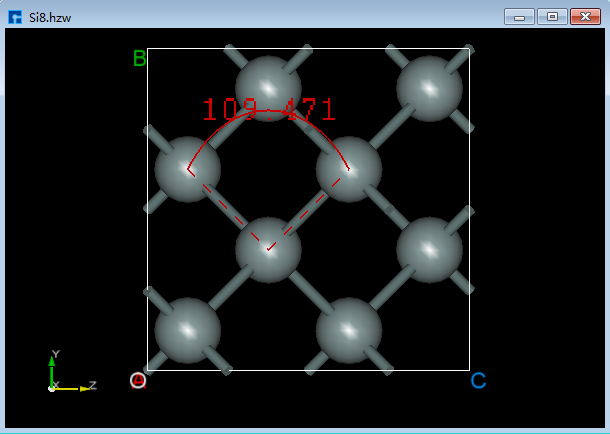
6.3.3. 测量3个原子之间的夹角¶
对于图6.3-1所示的晶体结构,先点击Toolbars上的 Angle 快捷图标,再通过鼠标分别选中结构中的3个原子则可测量选中的3个原子之间的夹角如图6.3-2(c)所示。